Методы исследование свойств сырья и продуктов питания
Нижняя граница определяемых содержаний (Сн) – это наименьшее значение определяемого содержания, ограничивающее интервал определяемых содержаний.
Верхняя граница определяемых содержаний (Св) – это наибольшее значение определяемого содержания, ограничивающее интервал определяемых содержаний.
(Сн) и (Св) обычно представляют собой массовую долю определяемого компонента в исследуемом продукте, а не в растворе.
Предел обнаружения (Смин) – наименьшее содержание, при котором по данной методике можно обнаружить присутствие определяемого компонента с заданной доверительной вероятностью.
Метод или методика анализа дают лишь тогда правильный результат, когда он свободен от систематических погрешностей. Систематические погрешности могут возникать на любом этапе аналитического процесса и по разным причинам. Задача освобождения результатов измерения от систематических погрешностей требует глубокого анализа всей совокупности данных измерений. Например, наиболее вероятным источником систематических погрешностей фотометрических измерений могут служить недостаточная представительность состава отобранной аналитической навески, погрешности в подготовке аналитической навески к фотометрическим измерениям, погрешности градуировки весов, мерной посуды, шкал спектрофотометров, несоответствие составов анализируемых и стандартных растворов, по которым строились градуировочные графики. Одной из часто встречающихся в физико-химических методах анализа причин систематических погрешностей является неправильное градуирование, в частности, построение градуировочных графиков на основе неподходящих градуировочных проб.
Воспроизводимость и сходимость результатов анализа определяются разбросом повторных результатов анализа относительно их среднего значения и обусловливаются наличием случайных погрешностей.
Сходимость характеризует рассеяние результатов при фиксированных условиях выполнения эксперимента; воспроизводимость – при варьировании этих условий. В первом приближении можно считать, что стандартное отклонение сходимости в 1,4–1,5 раза меньше стандартного отклонения внутри лабораторной воспроизводимости. Ввиду наличия такой простой связи между характеристиками сходимости и воспроизводимости в дальнейшем будет идти речь лишь о воспроизводимости как более общепринятом в литературе понятии.
Чем больше значение аналитических и инструментальных случайных погрешностей, тем менее точен анализ. Воспроизводимость характеризуется значением стандартного отклонения (S) или относительного стандартного отклонения (Sr).
Вольтамперметрические методы анализа пищевых продуктов
Методы анализа, основанные на расшифровке поляризационных кривых (вольтамперограмм), получаемых в электролитической ячейке с поляризующимся индикаторным электродом и неполяризующимся электродом сравнения, называют вольтамперометрическим. Вольтамперограмма позволяет одновременно получить качественную и количественную информацию о веществах, восстанавливающихся или окисляющихся на микроэлектроде (деполяризаторах), а также о характере электродного процесса.
В качестве поляризующегося микроэлектрода часто применяют ртутный капельный электрод, а сам метод называют в этом случае полярографией, следуя термину, который предложил Я. Гейровский, разработавший этот метод в 1922 г.
При небольшом потенциале катода сила тока сначала медленно увеличивается с возрастанием потенциала – это так называемый остаточный ток, его значение имеет порядок 10-7 А. По достижении потенциала восстановления на катоде начинается разряд ионов, определяемый диффузией, и сила тока резко возрастает, а затем становится постоянной – это предельный диффузионный ток.
Принципиальная схема полярографической установки приведена на рисунке 1.
Полярографическая установка включает в себя резервуар с ртутью, соединённый шлангом с капилляром, погруженным в анализируемый раствор. Электродом сравнения может служить слой донной ртути. В настоящее время, однако, чаще применяют обычные электроды сравнения – каломельный или хлоридсеребряный.
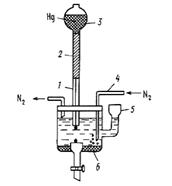
Рис. 1. Простейшая полярографическая ячейка:1 – стеклянный капилляр; 2 – полиэтиленовый шланг; 3 – груша с металлической ртутью; 4 – стеклянная трубочка с оттянутым концом для ввода азота; 5 – воронка для смены раствора;
6 – донная ртуть (Hg-анод)
Зависимость тока I от приложенного напряжения Е при обратимом электродном процессе передается уравнением полярографической волны:
Е = Е1/2 + (R T / n F) ln (Id – I) / I, (1)
Где Е1/2 – потенциал полуволны; Id – диффузионный ток.
При I = Id / 2 уравнение (1) переходит в
Е = Е1/2. (2)
Это соотношение показывает независимость потенциала полуволны от тока и, следовательно, от концентрации восстанавливающегося иона. Потенциал полуволны является, таким образом, качественной характеристикой иона в растворе данного фонового электролита, и определение потенциала полуволны составляет основу качественного полярографического анализа.
Количественный полярографический анализ основан на уравнении Ильковича, которое связывает диффузионный ток Id с концентрацией иона с и рядом других величин:
Id = 605 z D1/2 m 2/3 t1/6 c (3)
где z – заряд иона; D – коэффициент диффузии; m – масса ртути, вытекающей из капилляра за 1 с, мг; t – время образования капли (периода капания), с.
В практике количественного полярографического анализа коэффициент пропорциональности межу концентрацией вещества и силой диффузионного тока обычно устанавливают с помощью стандартных растворов. При постоянных условиях полярографирования D, m, и t постоянны, поэтому уравнение (3) переходит в
Id = k c. (4)
При анализе некоторых систем, для которых применимость уравнения (4) установлена вполне надежно, часто используют менее трудоемкий метод стандартных растворов. Так же широко распространен в количественной полярографии и метод добавок.
Особое место в полярографическом анализе занимает амперометрическое титрование.
Амперометрическое титрование представляет собой разновидность полярографического метода анализа. Амперометрическое титрование проводится следующим образом: часть исследуемого раствора помещают в электролизер, снабженный индикаторным электродом и электродом сравнения. Между электродами устанавливают напряжение на 0,3 – 0,5 В больше потенциала полуволны (или редокс-потенциала) исследуемого вещества и приступают к титрованию. В процессе титрования отмечают показания гальванометра, на основании результатов строят кривую амперометрического титрования, откладывая на оси ординат показания гальванометра, а на оси абсцисс – объем титранта. Точка перегиба соответствует объему титранта в точке эквивалентности. Содержание определяемого вещества вычисляют по объему титранта, израсходованному в точке эквивалентности. Концентрация титранта должна превышать концентрацию раствора титруемого вещества в 10–15 раз.
При амперометрическом титровании индикаторными электродами могут быть ртутный капельный электрод, платиновый вращающийся и другие электроды. В качестве электродов сравнения применяют насыщенный каломельный, хлорсеребряный и другие электроды.