Инновационный путь развития технологии создания новых лекарственных средств
При моделировании методами молекулярной динамики или Монте-Карло интересующее нас свойство системы большого числа молекул вычисляется через статистические средние по положениям и движениям молекул. Как и в методах молекулярной механики, здесь также необходимо перечислить все частицы системы и задать потенциалы межчастичных взаимодействий. Однако в отличие от молекулярной механики в данных подходах области задания межчастичных потенциалов взаимодействия должны быть достаточно протяженными, и они не должны ограничиваться малыми смешениями от положений равновесия. Это накладывает существенно более высокие требования на способы расчета потенциалов.
Практически всегда уравнения, связывающие молекулярные параметры и свойства вещества, то есть макроскопические свойства, решаются численно, а эффективность решения существенно зависит от мощности используемых компьютеров. На рисунке 2 показаны схемы двух методик: Монте-Карло и молекулярной динамики, применяемых в компьютерных экспериментах. В обоих случаях задаются число молекул N, объем V, доступный для движения молекул, накладываются те или иные граничные условия, предписывается потенциал межмолекулярного взаимодействия U. В методе Монте-Карло обычно независимыми переменными, сохраняющими постоянные значения при моделировании, выбираются N, V и температура Т. Молекулы двигаются случайным образом в соответствии с предписаниями генератора случайных чисел, и каждое новое расположение либо принимается, либо отбрасывается с вероятностью, определенной по закону
![]() ,
,
где k — константа Больцмана.
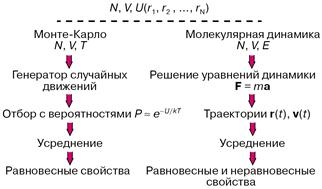
Рисунок 2 – Схема расчетов методами Монте-Карло и молекулярной динамики
При моделировании в рамках молекулярной динамики положения r(t) и скорости v(t) каждой частицы в момент времени t определяются как решения системы уравнений классической механики (уравнений Ньютона) либо уравнений, в которых к силам F задаваемым потенциалом U, добавляются так называемые случайные силы. Макроскопические свойства рассчитываются при усреднении по положениям и скоростям молекул.
Как уже упоминалось, число частиц при моделировании методами Монте-Карло и молекулярной динамики с помошью современных суперкомпьютеров может достигать колоссальных величин. Даже без суперкомпьютеров достаточно типичны численные эксперименты для значений N порядка десятков и сотен тысяч. Примеры успешного применения методов Монте-Карло и молекулярной динамики для моделирования равновесных составов смесей при постоянном давлении, фазовых равновесий, адсорбции на поверхности твердых тел, свойств жидкостей в микропорах и т.д. достаточно многочисленны. Этими же методами решаются задачи поиска устойчивых конформаций (поворотных изомеров) полимерных молекул, чрезвычайно важные для биохимических приложений [5,6].
Рассмотрим достаточно последовательную квантовую модель на примере бимолекулярной реакции типа
Х(i) + Y(j) → Х'(i') +Y'(j') + …
Здесь предполагается столкновение двух молекул X и Y, находящихся в состояниях i и j соответственно, которое приводит к продуктам реакции, то есть к молекулам X', Y', . в квантовых состояниях i', j', . Квантовая теория столкновений в принципе позволяет вычислить вероятности переходов между состояниями, отвечающими реагентам и продуктам, затем найти парциальные, то есть относящиеся к данным наборам квантовых чисел (здесь i, j, i', j', .), константы скорости. При усреднении по квантовым состояниям реагентов и продуктов можно оценить макроскопическую константу скорости соответствующей газофазной химической реакции как функцию температуры.
Полное осуществление этой программы в конкретных приложениях крайне затруднительно, даже если из предшествующих квантово-химических расчетов известна поверхность потенциальной энергии. Самой сложной стадией является численное решение уравнений квантовой теории столкновений с учетом перераспределения частиц, то есть как раз наиболее важная для химии стадия. Следует, однако, подчеркнуть исключительную важность научных исследований в этом направлении, поскольку они формируют каркас обшей теории, с которой сравниваются более простые модели. Кроме того, современные экспериментальные методы исследования динамики молекул позволяют измерить парциальные константы скорости и непосредственно сопоставить экспериментальные и теоретические результаты.
Более простые, а потому и более практичные способы вычисления констант скорости химических реакций получают обычно при определенных упрощениях полной квантовой модели. Так, начиная с 50-х годов проводятся компьютерные расчеты скоростей реакций методом классических траекторий. В этом методе, как и ранее, предполагается разделение электронной и ядерной подсистем, но в данном приложении необходимо знание поверхностей потенциальной энергии для достаточно широких интервалов межъядерных расстояний. Для расчета движений ядер, совместимых с данной потенциальной поверхностью, решают уравнения классической механики, а оценки констант скорости получают при сопоставлении числа траекторий, приводящих к реакции, с исходным числом траекторий при статистическом задании начальных условий.
На рисунке 3 изображены последовательные стадии вычислений методом классических траекторий. Реализация подобной схемы на современных компьютерах позволяет вычислять константы скорости реакций с существенно большим числом атомов, нежели при полностью квантовом описании [5,6].

Рисунок 3 – Схема расчетов констант скоростей химических реакций методом классических теорий
1.3 Определение биологической активности по модели
Для методов определения биологической активности вводится понятие о дескрипторах и QSAR. Молекулярный дескриптор – это числовые значения, характеризующие свойства молекул. Например, они могут представлять физико-химические свойства. Многие различные молекулярные дескрипторы описаны и применяются для различных целей. Они различаются по сложности, закодированной в них информации и сложности расчетов. Увеличение потребности в вычислительной технике увеличивается со сложностью расчетов.
Например, молекулярная масса имеет малое значение среди химических свойств, но зато быстро вычисляется. Дескрипторы основанные на квантово-химических расчетах имеют более важное значение для получения информации о химических свойств, но очень длительны по времени. Дескриптор может быть рассчитан из двухмерной и трехмерной модели химической структуры. Полученные дескрипторы обрабатываются и объединяются. Особое внимание заслуживают дескрипторы, которые описывают свойства молекул, а не замещают их. Такой вид дескрипторов является важной частью в разработке метода QSAR.
Большое распространение имеют математические и статистические модели. К таким моделям относятся модели методов QSAR(определяет количественные соотношения между структурой и активностью) и QSPR(определяет количественные соотношения между структурой и свойствами). Модель выполненная по методу QSAR должна быть разработана, как «эксперимент» и соответствовать точности реального эксперимента, для адекватного прогноза и для получения максимальной пользы от модели. В данном случае, чем больше первоначальных данных, тем точнее модель. Первым этапом является определение размера набора данных. Вторым этапом является корреляция дескрипторов. После определения набора данных и некоррелированных дескрипторов решается что именно должно быть включено в уравнения QSAR. Самый простой способ это использование автоматизированной процедуры. Полученные дескрипторы просчитываются через ряд уравнений и по полученным значениям определяют активность.