Методика обработки экспериментальных данных
Вся процедура обработки экспериментальных данных может быть разделена на два этапа. На первом производится первичная обработка сведений, полученных при проведении эксперимента по химическому равновесию, с целью определения значений констант равновесия. К этому же этапу относится и статистическая обработка данных, позволяющая провести выбраковку ошибочных сведений.
На втором этапе полученные значения констант равновесия подвергаются анализу c целью непосредственного определения роли энтальпийного и энтропийного факторов в равновесии позиционной изомеризации изучаемых веществ и выработки подходов к прогнозированию равновесия превращений родственных структур.
4.1. Первичная обработка экспериментальных данных
Первичная обработка результатов исследования химического равновесия основана на рекомендациях [38, 101-104] и включала в себя следующее.
Расчет отношений концентраций компонентов, характеризующих константы равновесия изучаемых реакций.
Анализ изученных отношений с целью установления момента достижения равновесия и включения в обработку только равновесных данных.
2. Исключение грубых ошибок внутри серии определений константы равновесия. Для каждой температуры исследования сериями считали опыты, различающиеся между собой либо составом исходной смеси, либо количеством катализатора. Отбраковку промахов в сериях проводили с использованием критерия – наибольшего по абсолютной величине нормированного выборочного отклонения:
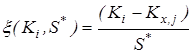 ,где
,где ![]() - значение константы равновесия;
- значение константы равновесия; ![]() - среднее арифметическое значение константы равновесия в серии j;
- среднее арифметическое значение константы равновесия в серии j;  - смещенная дисперсия
- смещенная дисперсия ![]() ;
; ![]() - число результативных определений константы равновесия в серии j.
- число результативных определений константы равновесия в серии j.
Процентные точки наибольшего по абсолютной величине нормированного выборочного отклонения заимствованы из работы [102].
3. Расчет среднего арифметического значения константы равновесия и дисперсии воспроизводимости в сериях после исключения грубых ошибок:
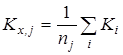
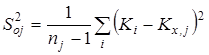
Сопоставление дисперсий воспроизводимости констант равновесия в сериях при одной температуре. Эта стадия дисперсионного анализа является весьма полезной, так как позволяет контролировать ошибки воспроизводимости, возникающие на всех этапах получения экспериментальной информации. Проверку равенства дисперсий воспроизводимости в сериях выполняли по двум критериям: Фишера – если число серий равнялось двум и Бартлетта – когда количество серий превышало два [38, 102]. Если нуль-гипотеза выполнялась, то дисперсию воспроизводимости вычисляли по следующей формуле:
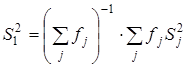 , где
, где ![]() .
.
Для всех изученных в данной работе превращений дисперсии воспроизводимости констант равновесия в сериях были однородны.
Расчет среднего значения константы равновесия ![]() для каждой температуры исследования:
для каждой температуры исследования:
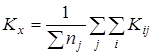
Проверку значимости расхождения средних значений констант равновесия в сериях. Для этого вычисляли дисперсию ![]() :
:
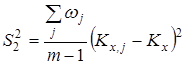
где m – число серий, wj – вес серии j, равный числу определений nj.
Величина ![]() характеризовала рассеяние значений
характеризовала рассеяние значений ![]() относительно
относительно ![]() . Число степеней свободы дисперсии
. Число степеней свободы дисперсии ![]() равнялось
равнялось ![]() . Проверку гипотезы равенства средних значений констант равновесия в сериях проводили с помощью распределения Фишера. Если нуль-гипотеза выполнялась, то вычисляли сводную дисперсию
. Проверку гипотезы равенства средних значений констант равновесия в сериях проводили с помощью распределения Фишера. Если нуль-гипотеза выполнялась, то вычисляли сводную дисперсию
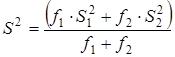
с числом степеней свободы ![]() . На этом обработка заканчивалась.
. На этом обработка заканчивалась.
Для всех исследованных реакций расхождения между серийными константами равновесия не превышали дисперсии воспроизводимости, что указывало на отсутствие систематических отклонений.
4.2. Анализ равновесных данных
Полученные значения констант равновесия анализировались с помощью подхода, основанного на последовательном исключении из исходной жидкофазной константы равновесия вкладов, обусловленных:
· межмолекулярными взаимодействиями,
· симметрией внешнего вращения молекул,
· вращением молекулы как целого,
· смешением конформеров,
· колебательным движением,
· вращением отдельных групп в молекулах реагентов.
Результатом подобного исключения является переход к газофазной константе равновесия ![]() , рассчитываемой следующим образом:
, рассчитываемой следующим образом:
![]() =exp(ln Kps- DS(or)/R - DS(mix)/R - DS(vib)/R - DS(ir)/R),
=exp(ln Kps- DS(or)/R - DS(mix)/R - DS(vib)/R - DS(ir)/R),
где ![]() - бессимметрийная газофазная константа равновесия реакции, DS(or)= SoII,g,(or) -SoI,g,(or); DS(mix)= SoII,g,(mix) -SoI,g,(mix); DS(vib)= SoII,g,(vib) -SoI,g,(vib); DS(ir)= SoII,g,(ir) -SoI,g,(ir).– соответственно, энтропийные вклады, обусловленные вращением молекулы как целого, смешением конформеров, колебательным движением и заторможенным вращением групп в молекуле, рассчитываемые как разность соответствующих энтропийных составляющих конечных (II) и исходных веществ (I).
- бессимметрийная газофазная константа равновесия реакции, DS(or)= SoII,g,(or) -SoI,g,(or); DS(mix)= SoII,g,(mix) -SoI,g,(mix); DS(vib)= SoII,g,(vib) -SoI,g,(vib); DS(ir)= SoII,g,(ir) -SoI,g,(ir).– соответственно, энтропийные вклады, обусловленные вращением молекулы как целого, смешением конформеров, колебательным движением и заторможенным вращением групп в молекуле, рассчитываемые как разность соответствующих энтропийных составляющих конечных (II) и исходных веществ (I).
Анализ констант равновесия проводился следующим образом.
Путем исключения вклада на симметрию молекул находилась бессимметрийная жидкофазная константа реакции 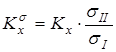 , здесь числа симметрии s формируются на основании как симметрии наружного вращения молекулы, так и симметрии внутреннего вращения тех групп, симметрия которых не может быть учтена при анализе энтропийного вклада, обусловленного внутренним вращением. Для систем, рассмотренных в данной работе, такими группами были фениленовые фрагменты.
, здесь числа симметрии s формируются на основании как симметрии наружного вращения молекулы, так и симметрии внутреннего вращения тех групп, симметрия которых не может быть учтена при анализе энтропийного вклада, обусловленного внутренним вращением. Для систем, рассмотренных в данной работе, такими группами были фениленовые фрагменты.
Путем снятия вклада на межмолекулярные взаимодействия рассчитывалась бессимметрийная газофазная константа равновесия реакции 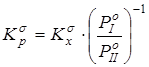 . Давления насыщенного пара
. Давления насыщенного пара ![]() рассчитывались методом Ли-Кеслера [50] или по экспериментальным данным. Применение к расчету давлений насыщенного пара методики, описанной в главе 2.1, позволяет обеспечить погрешность расчета не более 10% отн. для всех давлений, приведенных в данной работе. Вопросы расчета критических температур и давлений (Tc и Pc) изложены в главе 2, ацентрические факторы (w) рассчитывались по уравнению Ли-Кеслера [50].
рассчитывались методом Ли-Кеслера [50] или по экспериментальным данным. Применение к расчету давлений насыщенного пара методики, описанной в главе 2.1, позволяет обеспечить погрешность расчета не более 10% отн. для всех давлений, приведенных в данной работе. Вопросы расчета критических температур и давлений (Tc и Pc) изложены в главе 2, ацентрические факторы (w) рассчитывались по уравнению Ли-Кеслера [50].