Синтез 2,2-диэтоксииндандиона
3.1 Синтез индандиона-1,3
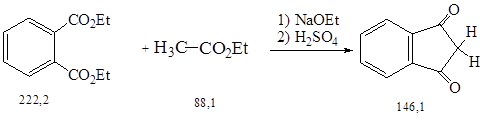
Синтез осуществляли по методике 2.1.
А. В трехгорлую колбу емкостью 500 мл с механической мешалкой, обратным холодильником и капельной воронкой поместили 100 г (0,45 моль) диэтилового эфира фталевой кислоты и 20 г (0,85 моль) мелконарезанного натрия.
Затем при перемешивании и нагревании прикапывали в течение 1 ч смесь 2,0 г (0,04 моль) этанола и 98,0 г (1,11 моль) этилацетата. После этого реакционную смесь кипятят с обратным холодильником еще 6 ч.
Полученную желтую суспензию охладили, добавили 50 мл безводного эфира и отфильтровали на воронке Бюхнера. Остаток на фильтре отсосали и высушили на воздухе. Выход натриевой соли этилового эфира 1,3-диоксоиндан-2-карбоновой кислоты составил 74,3 г (69 %).
Б. 2 л дистиллированной воды нагрели до кипения в стакане на 5 л и порциями при энергичном перемешивании внесли натриевую соль, полученную по методике А. Полученный раствор охладили до 70 0С и после окончания выделения диоксида углерода к полученному красному раствору прикапали при перемешивании 400 мл водного раствора серной кислоты (3:1).
Затем реакционную смесь охладили на бане со льдом до 15 0С, отсосали выпавший индандион-1,3 на воронке Бюхнера и после высушивания его в вакууме при 12 мм рт. ст. (Т=50 0С). Выход продукта составил 43,6 г (95 % в пересчете на натриевую соль) сырого желтого в-дикетона с Тпл. = 125–127 0С. Продукт перекристаллизовали растворением при нагревании в смеси 80 мл диоксана и 50 мл бензола (осторожно!) с добавлением 35 мл петролейного эфира (40–600С), а затем выделили в виде желтых игл.
Результаты анализа полученного вещества:
Тпл. = 129–131 0С
ИК (KBr): 1740, 1705 см-1 (С=О).
3.2 Синтез п-толуолсульфонилазида
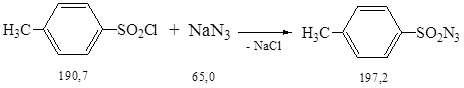
Синтез осуществляли по методике 2.2.
К раствору 95,2 г (0,50 моль) п-толуолсульфонилхлорида (предварительно перекристаллизованного из эфира) в 150 мл ацетона прикапывали при перемешивании раствор 35,7 г (0,55 моль) азида натрия в 100 мл воды и следили, чтобы температура реакционной смеси не поднималась выше 25 0С. Через 1,5 ч добавили еще 1 л воды и отделили органическую фазу на делительной воронке. Затем органическую фазу смешали с 250 мл воды и выделившийся тозилазид промыли водой (3 * 100 мл) и высушили над Na2SO4. При отсасывании получили маслянистый п-тозилазид с Тпл. = 5 0С, который затем полностью закристаллизовали. Выход твердого п-тозилазида составил 81 г (82 %).
Результаты анализа полученного вещества:
Тпл. = 5 0С
ИК (пленка): 2130 (N3), 1370, 1170 см-1 (SO2).
3.3 Синтез 2-диазоиндандиона-1,3
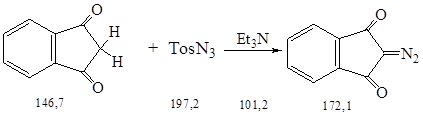
Синтез осуществляли по методике 2.3.
Суспензию 10,0 г (0,07 моль) тонкоизмельченного, охлажденного до –12 0С индандиона-1,3 в 60 мл безводного этанола прибавили при перемешивании к 9,0 г (0,09 моль) безводного триэтиламина; при этом температура поднимается до 4 0С. После повторного охлаждения к красно-коричневому раствору прилили 20,2 г (0,10 моль) п-тозилазида и смесь перемешали при охлаждении еще 1 ч.
Смесь отфильтровали, остаток на фильтре промыли охлажденным до –100С этанолом и после перекристаллизации из этанола получили 7,3 г (62 %) диазокетона в виде желтых игл.
Результаты анализа полученного вещества:
Тпл. = 147 0С
ИК (KBr): 2130, 2115 (С=N2), 1675 см-1 (С=О).
3.4 Синтез трет-бутилгипохлорита
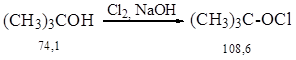
Синтез осуществляли по методике 2.4.
В двухгорлую круглодонную колбу объемом 250 мл поместили 100 мл NaOH (10 %) и прикапывали при перемешивании и охлаждении 7,4 г трет-бутанола. Затем смесь хлорировали около 30 мин., контролируя температуру, которая не должна подниматься выше 100С. Образовавшееся желтое масло отделили на делительной воронке и промыли холодной водой. Получили 5 г (46%) продукта с Ткип. = 77–78 0С.
Необходимый для синтеза хлор получили путем взаимодействия твердого перманганата калия с концентрированной соляной кислотой по методике, описанной в [7]. К 10 г твердого KMnO4, помещенного в колбу с отводом, прибавили по каплям из капельной воронки НСlконц. Поток полученного хлора осушали последовательно двумя промывалками с концентрированной серной кислотой, колонкой с безводным хлоридом кальция и колонкой с оксидом фосфора. После этого поток сухого хлора ввели в колбу со смесью гидроксида натрия и трет-бутанола (перед промывалками с серной кислотой необходимо поставить пустую обратную промывалку во избежании попадания серной кислоты в сосуд с перманганатом калия). Выход в атмосферу из колбы был защищен трубкой с безводным хлоридом кальция.
3.5 Синтез 2,2-диэтоксииндандиона-1,3
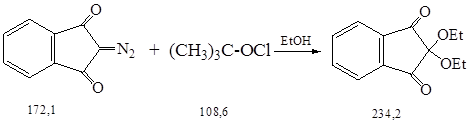
Синтез проводили по методике 2.6.
К суспензии 7,0 г (0,04 моль) тонкоизмельченного 2-диазоиндандиона-1,3 (см. п. 3.3.) в 60 мл этанола прикапали при перемешивании и охлаждении льдом 4,7 г (0,04 моль) трет-бутилгипохлорита (см. п. 3.4.) с такой скоростью, чтобы температура реакционной смеси не превышала 15 0С.
Вскоре из прозрачного раствора выпадают желтые кристаллы, которые через 30 мин. отсосали. Концентрирование маточного раствора при 30 0С/12 мм рт. ст. дало еще некоторое количество продукта. После перекристаллизации всех фракций из этанола получили 6,9 г (72 %) бледно-желтого кеталя с Тпл. = 84 0С.
Результаты анализа полученного вещества:
Тпл. = 84 0С
ИК (KBr): 1750, 1720 см-1 (С=О).
4. Обсуждение результатов
В результате проделанной работы был осуществлен ретросинтетический анализ и синтезирован 2,2-диэтоксииндандион-1,3 по следующей схеме:
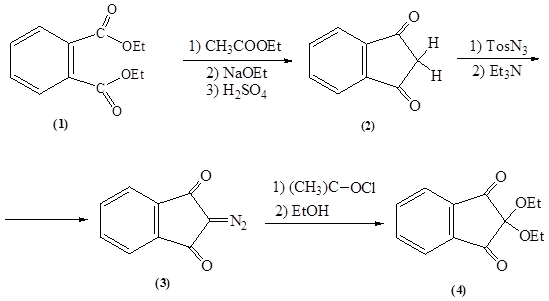
1 стадия: реакция образования пятичленного цикла сложноэфирной конденсацией этилового эфира уксусной кислоты с диэтиловым эфиром фталевой кислоты в присутствии сильного основания. Дальнейший гидролиз в-кетоэфира и декарбоксилирование образовавшейся в-кетокарбоновой кислоты.
Выход: 65 %.
2 стадия: реакция переноса диазогруппы путем взаимодействия индандиона-1,3 с п-толуолсульфонилазидом в присутствии основанияю
Выход: 62 %.
3 стадия: реакция гидролиза 2-диазоиндандиона-1,3 в присутствии окислителя с образованием 2,2-диэтоксииндандиона-1,3.
Выход: 72 %.
5. Выводы
1. На основании литературных данных был спланирован синтез 2,2-диэтоксииндандиона-1,3.
2. Освоена методика ретросинтетического анализа.
3. Был проведен трехстадийный синтез 2,2-диэтоксииндандиона-1,3, в результате которого выход целевого продукта составил 72 %. Полученное вещество было охарактеризовано и подтверждено данными ИК-спектроскопии.
4. Выходы веществ на всех стадиях были меньше, чем по литературным методикам, что было связано, по-видимому, с недостаточной очисткой исходных реагентов и потерями при очистке полученных веществ.