Специфика прогнозирования энтальпии образования ароматических органических соединений
|
C3-C3 |
10,204 |
1 |
Cl-F |
13,279 |
4 | ||||
|
C4-C4 |
93 |
I-I |
9,521 |
1 | |||||
|
C1-Ph |
5,962 |
1 |
(Nb-C1)орто |
-7,63 |
7 | ||||
|
(Nb-C1)пара |
-3,71 |
4 |
* - значения ряда парциальных вкладов и поправок для расчета DfH0g, 298 скорректированы по сравнению с первой редакцией пособия в связи с увеличением представительности выборки веществ, участвующих в определении параметра.
Дело в том, что различие в энергиях конформеров, обусловленных взаимной ориентацией атомов и групп соседних заместителей в ароматическом ядре, оказывается достаточным, чтобы учесть этот фактор при формировании аддитивной схемы. Суть вопроса становится понятной при конформационном анализе соединений с относительно несложным строением молекул, например метилбензолов (МБ), проведенного с помощью программы Gaussian 03W методом B3LYP/6-311G++2d,2p. Указанный метод дает для о-ксилола два сосуществующих конформера (“А” и “В”, рис. 1.2)
Один из них (“А”) характеризуется взаимным транс-расположением атомов водорода метильных групп, лежащих в плоскости ароматического ядра, в другом (“В”) атомы водорода метильных групп имеют шахматную ориентацию по отношению друг к другу. Различие в энергиях этих конформеров по результатам расчета составляет для ![]() 5,2 кДж/моль. Очевидно, что при таком соотношении в энергиях о-ксилол представлен при 298 К преимущественно конформером “А”, и эффект взаимодействия заместителей (орто-эффект типа “метил-метил” или C1-C1(транс-“Н-Н”)) для
5,2 кДж/моль. Очевидно, что при таком соотношении в энергиях о-ксилол представлен при 298 К преимущественно конформером “А”, и эффект взаимодействия заместителей (орто-эффект типа “метил-метил” или C1-C1(транс-“Н-Н”)) для ![]() составляет 2,76 кДж/моль. Но очевидно также и то, что указанное расположение метильных групп возможно только для двух соседних заместителей, третий и последующие заместители уже не могут иметь транс-ориентации атомов водорода. Выполненный нами конформационный анализ показал, что для 1,2,3-триМБ энергетически наиболее выгодным конформером является “С” (рис. 1.3), который для двух соседних групп имеет транс-ориентацию атомов водорода, лежащих в плоскости ароматического ядра, а третья метильная группа подстроена к соседней “шахматно”. Таким образом, суммарный эффект взаимодействия заместителей, дестабилизирующий молекулу 1,2,3-триМБ, составляет для
составляет 2,76 кДж/моль. Но очевидно также и то, что указанное расположение метильных групп возможно только для двух соседних заместителей, третий и последующие заместители уже не могут иметь транс-ориентации атомов водорода. Выполненный нами конформационный анализ показал, что для 1,2,3-триМБ энергетически наиболее выгодным конформером является “С” (рис. 1.3), который для двух соседних групп имеет транс-ориентацию атомов водорода, лежащих в плоскости ароматического ядра, а третья метильная группа подстроена к соседней “шахматно”. Таким образом, суммарный эффект взаимодействия заместителей, дестабилизирующий молекулу 1,2,3-триМБ, составляет для ![]() 7,63 кДж/моль (2,76+4,87), а величину 2,11 кДж/моль, равную 7,63-2·2,76 кДж/моль, можно было бы воспринять в рамках схемы Кокса-Пилчера [2] как дополнительный “тройной” эффект (сверх удвоенного орто-эффекта). Однако указанная ситуация в группе метилбензолов реализуется только в случае 1,2,3,5-тетраМБ, т.е. носит довольно частный характер. Расчет показывает, что для 1,2,3,4-тетраМБ и пента-МБ две группы транс-ориентированы, а остальные шахматно подстроены. В случае гекса-МБ все заместители имеют шахматную ориентацию.
7,63 кДж/моль (2,76+4,87), а величину 2,11 кДж/моль, равную 7,63-2·2,76 кДж/моль, можно было бы воспринять в рамках схемы Кокса-Пилчера [2] как дополнительный “тройной” эффект (сверх удвоенного орто-эффекта). Однако указанная ситуация в группе метилбензолов реализуется только в случае 1,2,3,5-тетраМБ, т.е. носит довольно частный характер. Расчет показывает, что для 1,2,3,4-тетраМБ и пента-МБ две группы транс-ориентированы, а остальные шахматно подстроены. В случае гекса-МБ все заместители имеют шахматную ориентацию.
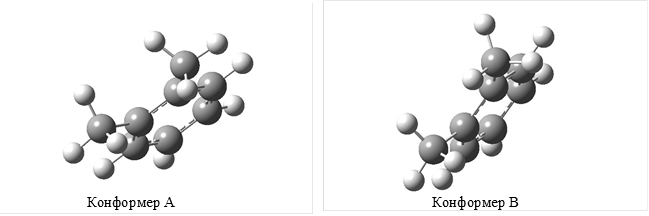
Рис. 1.2. Конформеры о-ксилола
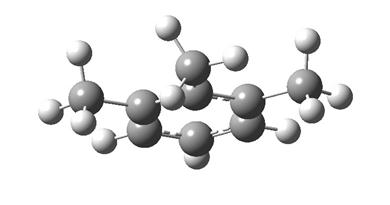
Рис. 1.3. Конформер C о-ксилола
На основании изложенного весь калориметрический материал для метилбензолов мог быть использован для определения всего двух поправок на орто-взаимодействие заместителей типа C1-C1(транс-“Н-Н”) и C1-C1(шахм.-“Н-Н”), значения которых приведены в табл. 1.11 и которые следует применять при прогнозировании ![]() целенаправленно в соответствии с взаимной ориентацией соседних групп.
целенаправленно в соответствии с взаимной ориентацией соседних групп.
Полагаем, что приведенный анализ и уровень значений орто-эффектов (табл. 1.11) предостережет от возможных ошибок при прогнозировании энтальпий образования веществ с более сложным строением молекул. Информация, представленная в табл. 1.11, свидетельствует о том, что спектр определенных орто-эффектов не охватывает всех возможных сочетаний взаимодействия алкильных заместителей и требуется пополнение экспериментальной базы. Для веществ со значительной степенью насыщения ароматического ядра алкильными заместителями различного строения приведенный набор параметров явно недостаточен. Однако детальный конформационный анализ интересующих структур с использованием возможностей методов молекулярной механики в сочетании со сведениями табл. 1.11 дает в большинстве случаев вполне удовлетворительные результаты при их оценке.
В отношении класса алкилфенолов в настоящее время можно сказать следующее. Введение всего одного парциального вклада типа “Cb-OH” (для учета взаимодействия ОН-группы с ароматическим ядром) в набор параметров, вычисленных на основе сведений по алкилбензолам и алканам, позволяет вполне корректно прогнозировать ![]() различных моно-, ди- и триметилфенолов, а также неэкранированных фенолов с алкильными заместителями иного строения. Частично экранированные и пространственно-затрудненные фенолы требуют учета эффектов взаимодействия ОН-группы с соседними алкильными заместителями. В методе Бенсона для этой цели рекомендована одна поправка, равная 2,38 и 1,42 кДж/моль, в редакции [5] и [6] соответственно.
различных моно-, ди- и триметилфенолов, а также неэкранированных фенолов с алкильными заместителями иного строения. Частично экранированные и пространственно-затрудненные фенолы требуют учета эффектов взаимодействия ОН-группы с соседними алкильными заместителями. В методе Бенсона для этой цели рекомендована одна поправка, равная 2,38 и 1,42 кДж/моль, в редакции [5] и [6] соответственно.
Значения орто-эффектов (табл. 1.11), определенные нами на основе экспериментальных данных, существенно зависят от эффективных размеров заместителей и их взаимной ориентации. Причем в случае фенолов последнее обстоятельство еще более четко выражено, чем для алкилбензолов. На основании многочисленных спектроскопических исследований и результатов выполненного нами конформационного анализа алкилфенолов (АФ) с различным строением молекул была установлена достаточно строгая ориентация гидрокси-групп по отношению как к плоскости ароматического ядра (практически всегда находится в плоскости), так и к соседнему алкильному заместителю. В соответствии с этим рекомендованы значения различных орто-эффектов, которые приведены в табл. 1.11 и предназначены для расчета ![]() с учетом следующего.
с учетом следующего.