Прыжки по хромосоме
Введение
В последнее десятилетие мы стали свидетелями целой серии ошеломляющих успехов в области молекулярной биологии. Разработка надежных методов клонирования, секвенирования и анализа экспрессии эукариотических генов углубила наши представления о структуре и регуляции активности гена, сделала более понятными механизмы многих наследственных болезней человека. В это же время быстро развивались и достигли значительных успехов методы картирования человеческих генов.
До недавнего времени существовал некий «провал» в области размеров хромосомных сегментов от 100 до 5000 т. п.н.; для них не имелось адекватных методов исследования. Такое положение значительно усложняло интерпретацию данных по картированию, полученных методами генетики соматических клеток и с помощью генетического анализа. Сопряжение таких данных с информацией, полученной на молекулярном уровне, стали называть «обратной генетикой».
В последние годы было предложено несколько подходов, позволяющих вести исследования в этой новой области. В настоящей работе описан один из них – метод «прыжков по хромосоме». С его помощью удается клонировать последовательности ДНК, значительно удаленные на генетической карте от последовательностей, гомологичных используемому зонду. Излагаются методики создания библиотек «хромосомных прыжков» и клонов-связок. Обсуждены преимущества и недостатки описываемого метода.
1. Область применения метода
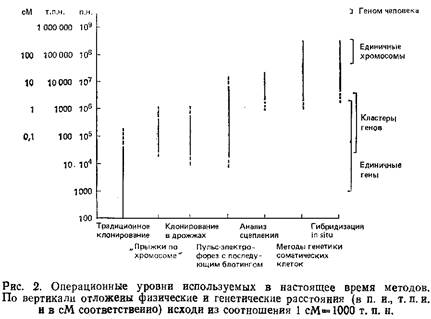
Рис. 2 дает представление о размерах хромосомных сегментов, в пределах которых «работают» различные современные методы генетических исследований. Ось ординат представляет собой логарифмическую шкалу физических расстояний, измеренных в парах нуклеотидов. На шкале приведены и значения генетических расстояний, измеряемые в сантиморганидах. 1 см приблизительно равна 108 п.н. Однако это соотношение нельзя считать универсальным, ибо зависимость между генетическим и физическим расстоянием на хромосоме имеет нелинейный характер, на нее могут оказывать влияние «горячие точки» рекомбинации. Наличие таких областей может привести к ситуации, когда сравнительно большому генетическому расстоянию соответствует небольшой отрезок на физической карте. В то же время в геноме существуют участки, рекомбинация в которых маловероятна, а это приводит к обратной ситуации. Как показано на рис. 2, классические методы молекулярной генетики хорошо работают на последовательностях длиной до 50 г.п.н., что соответствует максимальному размеру вставки в космидный вектор. Участки большей длины можно клонировать путем «прогулки по хромосоме», когда, используя уже клонированные последовательности, геномную библиотеку скринируют с целью получения перекрывающихся клонов. Таким способом удаётся анализировать последовательности длиной до нескольких сотен т. п.н. Однако, в некоторых случаях эта процедура может занять очень много времени. Так будет, если какие-то участки практически не перекрываются из-за наличия протяженной области повторяющихся последовательностей или последовательностей ДНК, которые не удается ввести в стандартные векторы. Если известно, что интересующий нас ген находится на расстояний нескольких сотен т. п.н. от используемого клона, то применение метода «прогулки по хромосоме» весьма проблематично.
На противоположном конце спектра работают методы генетики соматических клеток, гибридизация in situ, анализ генетического сцепления; их разрешающая способность ограничена 1000–5000 т. п.н. И наконец, середине шкалы соответствуют три метода, позволяющие использовать данные картирования для поиска специфических молекулярных нарушений. Это пульс-электрофорез, «прыжки по хромосоме» и клонирование в клетках дрожжей.
Важность этих подходов в том, что они дают ключ к пониманию молекулярных основ целого ряда генетических нарушений, для которых неизвестна функция кодирующих их генов. Известны наследственные болезни человека, связанные с аномалиями отдельных генов, такие, например, как муковисцидоз, болезнь Гентингтона, нейрофиброматоз. Наследование их происходит строго в соответствии с менделевскими правилами, они имеют четкие фенотипические характеристики, однако нормальные функции генов, кодирующих эти заболевания, не определены. Анализ сцепления с использованием полиморфных ДНК-маркеров позволил картировать соответствующие гены в специфических хромосомах человека, что в свою очередь, значительно увеличило возможности пренатальной и пресимптоматической диагностики. Чтобы до конца разобраться в природе этих заболеваний и предложить адекватные методы их лечения, необходим молекулярно-биологический метод, работающий в области больших молекулярных размеров. Именно таким методом и являются «прыжки по хромосоме».
2. Стандартные библиотеки «прыжков»
2.1 Типы «прыжков»
Следует разграничить понятия стандартных геномных библиотек для «прыжков по хромосоме» и специфических библиотек. В первом случае библиотеки создаются таким образом, что начинать движение вдоль хромосомы отмеренными прыжками можно в принципе с любой точки генома. Специфические библиотеки состоят из клонов, позволяющих осуществлять прыжки от одного редко встречающегося сайта рестрикции, например Notl, к последующему такому же сайту. Типы прыжков схематически изображены на рис. 3. Очевидно, что способы создания этих библиотек несколько различаются. С технической точки зрения труднее получать библиотеки первого типа, так как они должны содержать репрезентативную выборку последовательностей генома. Обычно для такой библиотеки требуется 3*108 клонов. Тогда можно быть твердо уверенным, что с ней можно работать, начиная от любой стартовой точки на хромосоме.
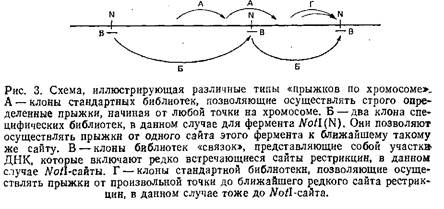
Для создания же полных специфических библиотек требуется всего лишь 10000-20000 клонов, так как количество независимых клонов эквивалентно числу рестрикционных фрагментов, полученных при использовании данного фермента рестрикции. Так, например, для рестриктазы Notl, которая отщепляет приблизительно по 1000 т. п.н. в геноме человека, таких фрагментов должно быть всего лишь около 3000. Поэтому библиотеку из 10000 клонов для прыжков по Not сайтам можно считать практически полной. Очевидным недостатком таких библиотек является то, что их нельзя использовать, когда стартовая точка прыжка не примыкает к редко встречающемуся сайту рестрикции. К сожалению, это довольно частое явление, препятствующее быстрому распространению данного метода. Если же все-таки удается идентифицировать клон, примыкающий к редко встречающемуся сайту рестрикции, использование специфических библиотек для «прыжков по хромосоме» может оказать неоценимую помощь.
Очень эффективным могло бы оказаться создание геномной библиотеки третьего типа, имеющей в своем составе клоны, соединяющие редко встречающиеся сайты рестрикции с прилегающими случайными последовательностями ДНК генома. Имея такую библиотеку, мы могли бы начинать движение по хромосоме с любой стартовой точки, а при встрече с редким сайтом рестрикции пускать в ход специфическую геномную библиотеку. К сожалению, пока эта задача не решена и подходы к ее решению здесь обсуждаться не будут.