Контактное окисление и адгезия к стали полиэтиленовых покрытий
Ранее [1,2] было установлено, что адгезия ПЭ-покрытий, окисляющихся на каталитически неактивной подложке, является функцией не только температуры Т и времени окисления т, но и толщины покрытий h. Уменьшение адгезии, характеризуемой удельным сопротивлением отслаиванию покрытий f при увеличении т обусловлено в основном проникновением к подложке низкомолекулярных продуктов термоокислительной деструкции макромолекул наружного поверхностного слоя покрытий. При повышении Т существенную роль в снижении величины f играет также уменьшение степени окисления граничащего с подложкой слоя полимера, связанное с ускорением процесса окисления в поверхностном слое покрытий.
Окисление граничащего с подложкой слоя осуществляется в основном кислородом, диффундирующим в покрытие из окружающей среды [3,4], а в катализе окисления полиолефинов на металлах существенную роль играют металлосодержащие продукты контактных реакций окисляющегося полимера с металлом, диффундирующие в покрытие (в основном соли карбоновых кислот) [5—7]. Поэтому следует ожидать, что закономерности изменения величины f для покрытий на каталитически активной подложке могут быть иными, чем на каталитически неактивной.
Цель данной работы — изучение адгезии ПЭ-покрытий толщиной 100—4000 мкм, окисленных на каталитически активной подложке (стали) [4,8,9] при различных температурно-временных условиях в среде воздуха: Г=433-453К, т=0,03-10,8 кс.
На рис. 1 представлены данные по влиянию т при различных температурах окисления на величину f для покрытий различной толщины на стали. Зависимость f от т при постоянных h и Т описывается кривой с максимумом. При увеличении h покрытий значение т, соответствующее максимуму f на изотермической кривой, сначала уменьшается, а затем увеличивается (рис. 1; рис. 2, б, кривая 4), в то время как для покрытий на каталитически неактивной подложке (алюминий [5]) оно только увеличивается [1]. Вследствие этого зависимость величины f покрытий на стали от h для постоянных Т и τ описывается при малых значениях τ кривой с максимумом, при средних т кривой с двумя максимумами, при больших т кривой с максимумом (рис. 3, а, кривые 1—3).
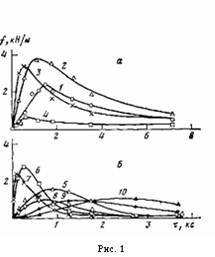
Рис. 1. Зависимость сопротивления отслаиванию ПЭ-покрытий на стали от времени их окисления т при 433 (а) и 453 К (б). Толщина покрытий 200 (1), 300 (5), 500 (2, 6), 800 (3, 7), 1000 (4, 8), 2000 (9) и 3000 мкм (10)
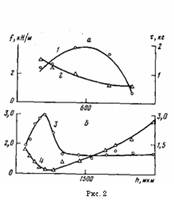
Рис. 2. Влияние толщины h ПЭ-покрытий на максимальные значения / (1, 3) для покрытий, окисленных при 433 (а) и 453 К (б), и время (2, 4), необходимое для достижения максимальной величины при 433 (а) и 453 К (б)
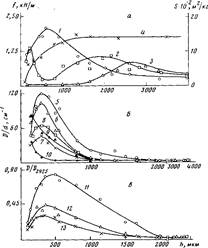
Рис. 3. Влияние толщины ПЭ-покрытий на стали, окисленных при 453 К в течение 0,6 (1), 1,2 (2, 10), 2,7 (S, 9) и 3,6 кс (3, 4, 5-7, 11-13), на сопротивление отслаиванию f (1—3), на площадь пика плавления S (4) (а), на накопление в граничащем со сталью слое солей карбоновых кислот (5, 8, 11), карбонильных (6, 9, 10, 12) и эфирных групп (7, 13) (б, в); б - данные, полученные методом ИК-спектроскопии пропускания для граничащего слоя толщиной 10 мкм; в — данные, полученные методом многократного нарушенного полного внутреннего отражения. Здесь и на рис. 5: D — оптическая плотность, d — эффективная толщина поглощающего слоя
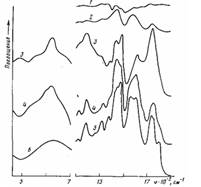
Рис. 4. ИК-спектры пропускания (1, 5) граничащего со сталью ело» толщиной 10 мкм (5) и остатков полимера на стали (1), а также ИК' спектры многократного нарушенного полного внутреннего отражения (2—4, 6) поверхности окисленной стали (6), стали, отслоенной от ПЭ-покрытия (2), и поверхности покрытия, контактировавшего со сталью, до (4) и после обработки в течение 7,2 кс раствором серной кислоты (3). Толщина покрытия 300 (1) и 500 мкм (2-5); время окисления при 453 К 0,6 (1); 1,8 (2); 3,6 кс (3-5)
При увеличении т для покрытий на стали в отличие от покрытий на алюминии f уменьшается после достижения максимума практически до нуля, а в слое, граничащем с подложкой, накапливаются соли карбоновых кислот — продукты контактных реакций карбоновых кислот, образующихся при окислении ПЭ с металлом. При понижении f с увеличением т уменьшается количество полимера, остающегося на подложке после ее отслаивания, и возрастает содержание на ней солей карбоновых кислот. При низких значениях f остающийся на подложке слой практически весь состоит из солей карбоновых кислот (полоса поглощения симметричных колебаний карбоксилат-аниона 1600 см-1, антисимметричных — 1440 см-1) (рис. 4, кривая 2). На отслоенной поверхности полимерного покрытия также фиксируются преимущественно соли карбоновых кислот (рис. 4, кривая 4). Следовательно, разрушение происходит по слою солей, а снижение f при увеличении т связано с их накоплением в зоне адгезионного контакта. Большое количество солей обнаруживается в слое толщиной 10 мкм, срезанном с поверхности отслоенного покрытия, бывшей в контакте с подложкой (рис. 4, кривая 5). Регистрируются они также в покрытиях, имеющих максимальные значения f (рис. 4, кривая 1). Кроме того, для отслоенного от подложки покрытия наблюдается сильное поглощение в области 500—700 см-1 (рис. 4, кривая 4), характерное для связи кислород — металл в карбоксилатах и оксидах железа (рис. 4, кривая 6). Оксиды железа в полимере могут образовываться вследствие окисления атомарного металла, появляющегося при распаде солей или при диффузии ионов и атомов металла в полимер. Ранее [1] было показано, что если к окисленному на каталитически неактивных подложках (алюминий, стекло) ПЭ-покрытию припрессовывать расплавленную неокисленную ПЭ-пленку, то значение f, снизившееся при увеличении τ, увеличивается вследствие перехода части низкомолекулярных продуктов окисления (карбоновых кислот и т. п.) из зоны адгезионного контакта в припрессованную пленку.
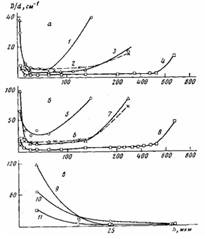
Рис. 5. Распределение по толщине h эфирных (а) и карбонильных групп (б), а также солей карбоновых кислот (в) для покрытий толщиной 200 (1, 5, 10), 300 (2, 3, 6, 7, 9) и 800 мкм (4, 8, 11), окисленных при 453 К в течение 3,6 кс на алюминии (2, 6) и на стали (1, 3, 4, 5, 7, 8, 9-11)
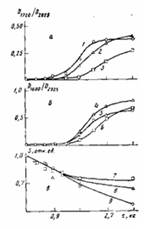
Рис. 6. Влияние времени окисления т при 453 К на накопление в граничащем со сталью слое ПЭ-покрытий толщиной 200 (1, 5, 9), 500 (2, 4, 8) и 800 мкм (3, 6, 7) карбонильных групп (а), солей карбоновых кислот (б) и изменение площади пика плавления S (в) на термограммах дифференциальной сканирующей калориметрии. Для зависимостей S—х толщина анализируемого слоя 10 мкм
Если проделать такую же операцию с покрытиями, окисленными на стали, то существенного восстановления значения f не происходит. Это также является свидетельством того, что причина спада величины f при окислении ПЭ-покрытий на стали заключается в накоплении в зоне адгезионного контакта малорастворимых в ПЭ солей карбоновых кислот.