Контактное окисление и адгезия к стали полиэтиленовых покрытий
Для условий, при которых значение f покрытий на стали с увеличением т падает (рис. 1, а), водостойкость адгезионных соединений увеличивается. Водостойкость повышается также при увеличении h покрытий, когда наблюдается возрастание концентрации солей в граничащем с подложкой слое. Однако такие адгезионные соединения ПЭ со сталью являются нестойкими в минеральных кислотах. Например, в водном растворе серной кислоты происходит самоотслаивание покрытий. Эти результаты легко объясняются, если учесть, что карбоксилаты железа нерастворимы в воде, но хорошо растворимы в серной кислоте и ее водных растворах. При растворении происходит реакция с образованием растворимых в воде сульфатов железа и карбоновой кислоты. Соли карбоновых кислот в отслоенных тонких покрытиях после их обработки в серной кислоте не обнаруживаются, а в толстых покрытиях их количество уменьшается (рис. 4, кривая 3). Как и следовало ожидать, одновременно увеличивается содержание карбоновых кислот в полимере, что характеризуется возрастанием интенсивности пика в области 1710—1720 см-1 (рис. 4, кривая 3). При этом уменьшается интенсивность поглощения в области 500—700 см-1. Последнее согласуется с предположением о том, что поглощение в указанной области связано с оксидами металла.
Карбоксилаты железа плохо растворимы в расплаве ПЭ и интенсивно накапливаются в граничащем с подложкой слое, при формировании Покрытий. Отметим, что аналогичные соли свинца, а также меди значительно лучше растворяются в расплаве ПЭ [5—7] и значение f для покрытий на свинце и на меди при увеличении т не уменьшается до нулевых значений [10,11]. При увеличении h толщина слоя покрытия, в котором регистрируются соли карбоновых кислот, уменьшается (рис. 5, в) вследствие локализации окисления в более тонком граничащем с подложкой слое покрытия (рис. 5, а, б, кривые 1, 3—8). Для ПЭ-покрытии на каталитически активной подложке в отличие от покрытий на каталитически неактивной подложке окисление наиболее интенсивно происходит в граничащем с подложкой слое, а также в поверхностном (наружном) слое (рис. 5, а, б). Для толстых покрытий окисление граничащего с подложкой и поверхностного слоев осуществляется независимо, а степень окисления граничащего с подложкой слоя меньше, чем у тонких покрытий (рис. 5, а, б, кривые 1, 4,5,8; рис. 6, а). При этом на определенной глубине от поверхности покрытия концентрация карбонильных групп резко уменьшается (рис. 5, б, кривая 8). По-видимому, на этой глубине концентрация кислорода в полимере недостаточна, чтобы преодолеть индукционный период окисления без каталитического воздействия подложки. Не исключено, что это связано с диффузией низкомолекулярных карбонилсодержащих продуктов окисления, образующихся в наружном слое. Для толстых покрытий, у которых окисление граничащего с подложкой и поверхностного слоев происходит независимо, растворимые в полимере низкомолекулярные продукты окисления, образующиеся в граничащем с подложкой слое полимера, могут диффундировать в среднюю неокисленную или малоокисленную часть покрытия. Эта часть покрытия для окисленного граничащего с подложкой слоя полимера может выполнять функцию «припрессованной пленки». При малых толщинах происходит взаимовлияние процессов окисления в граничащих с подложкой и в поверхностных слоях (рис. 5, а, б, кривые 1, 5). При этом концентрация карбонильных, эфирных и других кислородсодержащих групп в слое, граничащем с подложкой, уменьшается с уменьшением h покрытий при больших т (рис. 5, а, б; кривые 1,3,5,7), т. е. зависимость концентрации этих групп в граничащем с подложкой слое от толщины покрытия может описываться кривой с максимумом (рис. 3, б, в; кривые 6, 7,12,13; рис. 6, а). Аналогичной по характеру зависимостью описывается при этом накопление в граничащем с подложкой слое солей карбоновых кислот (рис. 3,6, в, кривые 5,8,11; рис. 6, б). Уменьшение концентрации кислородсодержащих групп и солей в граничащем с подложкой слое тонких покрытий на стали при уменьшении их толщины для постоянных Т и τ связано с участием каталитически активной подложки в окислении наружного поверхностного слоя покрытий. Соли карбоновых кислот лучше растворимы в частично окисленном ПЭ и регистрируются для более тонких покрытий, например, методом ИК-спектроскопии в более отдаленных от подложки слоях (рис. 5, в, кривая 10). При этом происходит более быстрое окисление поверхностного слоя (рис. 6, а, кривая 1), увеличивается окислительная сшивка макромолекул, способствующая аморфизации ПЭ, и раньше наступает характерный для окисления ПЭ-пленок на каталитически активных металлах (медь, свинец и др.) эффект автоингибирования окисления [5,12] (рис. 6, а, кривые 1,2). В результате уменьшается удельная (приходящаяся на единицу толщины), а также общая степень окисления покрытий (рис. 6, а, кривые 1,2; рис. 7). Чем выше степень окисления, тем больше образуется низкомолекулярных карбоновых кислот в покрытии. Следовательно, определенный вклад в накопление солей в граничащем с подложкой слое тонких покрытий могут вносить и карбоновые кислоты, диффундирующие из наружного слоя в зону адгезионного контакта. При окислении происходит аморфизация граничащего с подложкой слоя полимера (рис. 3, а, кривая 4; рис. 6, в). Если условия автоингибирования не достигнуты, то степень окисления ПЭ-покрытий, а также граничащего с подложкой слоя полимера уменьшается при увеличении h (рис. 3, б, кривые 9,10). Отметим, что основные изменения значения f происходят в условиях, когда автоингибирование еще не достигается (рис. 1, б; рис. 3; рис. 6, с).
Полученные данные показывают, что толщина покрытий существенно влияет на степень окисления граничащего с каталитически активной подложкой слоя, на накопление в нем солей карбоновых кислот и на адгезию покрытий к подложке. Для тонких покрытий при малых τ, когда автоингибирование окислительного процесса не достигнуто, увеличение толщины покрытия приводит к уменьшению скорости накопления карбонильных и эфирных групп в граничащем с подложкой слое (рис. 6, а, кривые 1,2; рис. 3, б, кривые 8,10) и к увеличению скорости накопления в нем солей(рис. 6, б, кривые 4,5; рис. 3, б, кривая 8). Так как спад величины f на зависимости f от τ обусловлен накоплением в зоне адгезионного контакта солей, смещение максимума f в область меньших т при увеличении h тонких покрытий связано с увеличением скорости накопления солей, а не с изменением степени окисления полимера, как можно было ожидать исходя из существующих представлений [8,9].
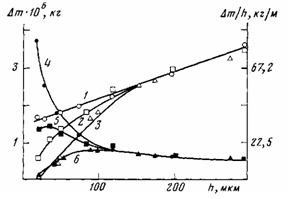
Рис. 7. Влияние толщины ПЭ-покрытий на алюминии (1—4), стали (2, 5) и меди (3, 6) на приращение массы Am (1—3) и удельное приращение массы Am/h (4-6), по данным ТТА, при окислении ПЭ-покрытий в иеизотермическях условиях
При этом зависимость максимальных значений f от h покрытий описывается кривой с максимумом (рис. 3, а, кривые 1,3), что можно объяснить уменьшением толщины окисленного граничащего с подложкой слоя полимера. Известно [13], что зависимость прочности соединений металлов с полимерами от толщины промежуточного тонкого окисленного полимерного слоя описывается кривой с максимумом. Определенную роль при этом может играть аморфизация граничащего с подложкой слоя полимера. Для толстых покрытий (1200 мкм и более) максимум величины f смещается в область больших значений τ при увеличении h (рис. 1, б, кривые 8—10), что связано, по нашему мнению, с влиянием толщины пленки на количество кислорода, проникающего в зону адгезионного контакта, т. е. с диффузионным ограничением контактного окисления.