Прогнозирование критической температуры. Алканы и алкены
Сложность прогнозирования критической (жидкость-пар) температуры органических веществ состоит в том, что Тс изменяются нелинейно с изменением числа углеродных атомов в молекуле даже в отдельно взятой гомологической группе (рис. 5.1.). Аддитивные методы для таких свойств оказываются неэффективными, поскольку нелинейность свойства сохраняется для значительного количества соединений при переходе от низших представителей гомологических групп к высшим. Это не позволит принять некоторое постоянное значение даже для парциального вклада, характеризующего гомологическую разность, т.е. вклад на СН2 группу.
Для таких свойств широко используются аддитивно-корреляционные методы, в которых вид корреляции ответственен за изменение свойства в гомологической группе, а аддитивная составляющая свойства передает его связь со строением молекул. Рассчитывать на успех в применении этих методов возможно только в случае одинаковых соотношений типа “значение свойства - количество углеродных атомов в любой гомологической группе”. Из рис. 5.1 следует, что для критических температур это условие также не выполняется.
Приблизиться к решению проблемы удалось, используя аддитивно-корреляционные методы с дополнительной опорой на родственное с критической температурой свойство вещества. В качестве такого свойства наилучшим образом выступает нормальная температура кипения (Tb). С одной стороны, предельно близка природа этих свойств, с другой - Tb наиболее полно по сравнению с другими физико-химическими свойствами подкреплены справочными данными. Именно Tb является опорным свойством в большинстве методов прогнозирования критических температур.
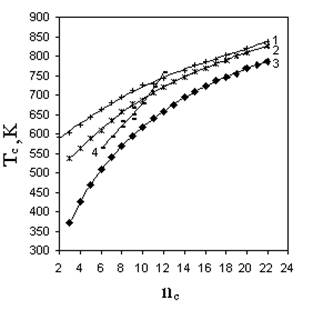
Р и с. 5.1. Зависимость критической температуры от числа углеродных атомов в молекуле:
1 - н-монокарбоновые кислоты; 2 – н-спирты;
3 – н-алканы; 4 – бензол - метилбензолы
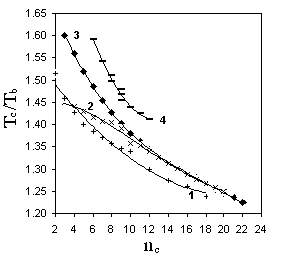
Р и с. 5.2. Зависимость Tc/Tb от числа углеродных атомов в молекуле:
1 - н-монокарбоновые кислоты;
2 – н-спирты; 3 – н-алканы;
4 – бензол - метилбензолы
Иллюстрацией того, что указанный прием позволяет несколько упростить задачу прогнозирования Tс, является рис. 5.2. Однако наряду с этим из рис. 5.2 следует, что использование Tb в качестве опорного свойства не гарантирует успеха при прогнозировании Tс на основе общих универсальных корреляций для соединений любых классов. Примером тому служит совершенно иной по сравнению с соединениями прочих приведенных на рис. 5.2 классов вид корреляции для первичных спиртов С3-С10.
Метод Джобака
Сохранив основу метода Лидерсена, Джобак ввел некоторые коррективы в вид корреляции и параметризацию аддитивных составляющих свойства. В результате погрешность метода для веществ с относительно высокой молекулярной массой уменьшилась (табл. 5.1).
Корреляция, рекомендуемая для прогнозирования критических температур, требует знания нормальных температур кипения вещества и имеет вид
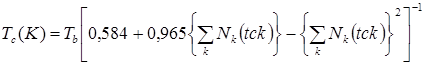 ,(5.2)
,(5.2)
где значения нормальной температуры кипения и критической температуры даны в градусах Кельвина.
Величины парциальных вкладов (tck) в критическую температуру приведены в табл. 5.3. В табл. 5.1, 5.5 приводятся результаты прогнозирования Тс методом Джобака для соединений различных классов, откуда следует, что явных преимуществ перед методом Лидерсена он все-таки не имеет. Учитывая то, что эти два метода достаточно просты в применении и в ряде случаев дают разнонаправленные отклонения, целесообразно использовать их совместно, чтобы таким образом несколько снизить погрешность в оценках критической температуры.
Следует отметить, что авторами сделана попытка увеличения точности прогноза этими методами. С этой целью был заменен вид корреляции, увеличена ее гибкость, и согласованы значения парциальных вкладов с полученной корреляцией. Однако принципиального улучшения результатов прогноза при этом не про изошло. Таким образом, возможности аддитивно-корреляционного подхода с параметризацией по атомам практически полностью использованы в методе Джобака.
Авторы сочли также целесообразным привести результаты прогнозирования Тс алканов другими методами (табл. 5.1), идеология которых не рассматривалась по следующим соображениям. Для ряда методов погрешности в оценках слишком велики уже для алканов; очевидно, что для соединений прочих классов они не станут меньше.
Метод Марреро-Пардилло дает для алканов хорошие оценки. Однако методу свойственны систематические отклонения, обусловленные избыточной жесткостью принятой авторами параболической корреляции. Аддитивная составляющая свойства формируется методом по связям аналогично тому, как это показано нами для энтальпий образования (глава 1). Использование метода по связям неизбежно повышает точность прогноза по сравнению с методами по атомам (Лидерсена и Джобака), но предъявляет серьезные требования к объему исходной информации.
Метод, основанный на индексах молекулярной связности Рандича
Поиск путей повышения точности прогноза критических температур заставил нас обратить внимание на метод, основанный на теории графов и индексах молекулярной связности Рандича [43]. Индексы Рандича наряду с другими дескрипторами более 25 лет с успехом применяются для прогнозирования различных свойств органических веществ [44-47]. Заметную роль в их практическом применении сыграли Кир и Халл [44-46]. Метод представляется нам достаточно привлекательным своей внутренней логикой. Суть метода состоит в следующем.
На основе кодовых чисел ![]() атомов в молекуле определяются индексы молекулярной связности
атомов в молекуле определяются индексы молекулярной связности ![]() . Индекс молекулярной связности первого порядка равен
. Индекс молекулярной связности первого порядка равен ![]() и отражает вклад в свойство валентно связанных атомов. Индекс молекулярной связности второго порядка характеризует вклад в свойство трех последовательно расположенных атомов в молекуле и равен
и отражает вклад в свойство валентно связанных атомов. Индекс молекулярной связности второго порядка характеризует вклад в свойство трех последовательно расположенных атомов в молекуле и равен ![]() . В формировании этого и последующих индексов принимают участие не только валентно связанные атомы, но и все те, через которые осуществляется “передача взаимодействия атомов”. При этом степень участия “атомов-посредников” оказывается зависимой от величины их кодовых чисел. Этот факт является, на наш взгляд, весьма сильной стороной метода.
. В формировании этого и последующих индексов принимают участие не только валентно связанные атомы, но и все те, через которые осуществляется “передача взаимодействия атомов”. При этом степень участия “атомов-посредников” оказывается зависимой от величины их кодовых чисел. Этот факт является, на наш взгляд, весьма сильной стороной метода.
Рандичем значения кодовых чисел были заданы [43]. Например, для первичного, вторичного, третичного и четвертичного атомов углерода были приняты соответственно значения 1, 2, 3 и 4. Киром и Халлом в редакции [44] значения ![]() рассчитываются на основе количества валентных электронов и общего их числа в атоме. Таким образом, в том и в другом случаях кодовые числа имеют постоянное значение для каждой разновидности атомов и не зависят от прогнозируемого свойства.
рассчитываются на основе количества валентных электронов и общего их числа в атоме. Таким образом, в том и в другом случаях кодовые числа имеют постоянное значение для каждой разновидности атомов и не зависят от прогнозируемого свойства.