Апоптоз - программируемая клеточная смерть
Каспаза-8 активирует каспазу второго эшелона (эффекторную каспазу): путем протеолиза из прокаспазы-3 образуется каспаза-3, после чего процесс, запущенный программой смерти, оказывается необратимым.
Каспаза-3 способна в дальнейшем к самостоятельной активации (автокатализу или автопроцессингу), активирует ряд других протеаз семейства каспаз, активирует фактор фрагментации ДНК, ведет к необратимому распаду ДНК на нуклеосомальные фрагменты. Так запускается каскад протеолитических ферментов,осуществляющих апоптоз.
2.Второй путь реализации программы ПКС.
В клетках, подвергшихся воздействию индуктора апоптоза, резко снижается мембранный потенциал (Dy)митохондрий . Падение Dy обусловлено увеличением проницаемости внутренней мембраны митохондрий вследствие образования гигантских пор . Разнообразны факторы, вызывающие раскрытие пор . К ним относятся истощение клеток восстановленным глутатионом, NAD(P)H, ATP и ADP, образование активных форм кислорода, разобщение окислительного фосфорелирования протонофорными соединениями, увеличение содержания Ca2+ в цитоплазме. Образование пор в митохондриях можно вызвать церамидом, NO, каспазами, амфипатическими пептидами, жирными кислотами . Поры имеют диаметр 2,9 нм, позволяющий пересекать мембрану веществам с молекулярной массой 1,5 кДа и ниже. Следствием раскрытия поры является набухание митохондриального матрикса, разрыв наружной мембраны митохондрий и высвобождение растворимых белков межмембранного объема . Среди этих белков – ряд апоптогенных факторов: цитохром с , прокаспазы 2, 3 и 9 , белок AIF (apoptosis inducing factor), представляющий собой флавопротеин с молекулярной массой 57 кДа [69].
Образование гигантских пор не является единственным механизмом выхода межмембранных белков митохондрий в цитоплазму. Предполагается , что разрыв наружной мембраны митохондрий может быть вызван гиперполяризацией внутренней мембраны. Возможен и альтернативный механизм, без разрыва мембраны, – раскрытие гигантского белкового канала в самой наружной мембране, способного пропускать цитохром с и другие белки из межмембранного пространства .
Высвобождаемый из митохондрий цитохром с вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) участвует в активации каспазы-9 .
APAF-1 – белок с молекулярной массой 130 кДа, содержащий CARD-домен (caspase activation and recruitment domain) образует комплекс с прокаспазой-9 в присутствии цитохрома с и dATP или АТР. Из этих субъединиц собираются жесткие, симметричные структуры, наподобие веера или пропеллера .APAF-1 играет роль арматуры, на которой происходит аутокаталитический процессинг каспазы-9 . Предполагается, что в результате зависимого от гидролиза dATP (или АТР) конформационного изменения APAF-1 приобретает способность связывать цитохром с (рис. 5). Связав цитохром с, APAF-1 претерпевает дальнейшее конформационное изменение, способствующее его олигомеризации и открывающее доступ CARD-домена APAF-1 для прокаспазы-9, которая тоже содержит CARD-домен. Так образуется конструкция, называемая тоже апоптосомой, с молекулярной массой > 1,3 млн дальтон, в составе которой – не менее 8 субъединиц APAF-1 . Благодаря гомофильному CARD-CARD-взаимодействию с APAF-1 в эквимолярном соотношении связывается прокаспаза-9, а затем прокаспаза-9 связывает прокаспазу-3. Пространственное сближение молекул прокаспазы-9 на мультимерной арматуре из APAF-1-цитохром-с-комплексов, по-видимому, приводит к межмолекулярному протеолитическому процессингу прокаспазы-9 с образованием активной каспазы-9. Зрелая каспаза-9 затем расщепляет и активирует прокаспазу-3. 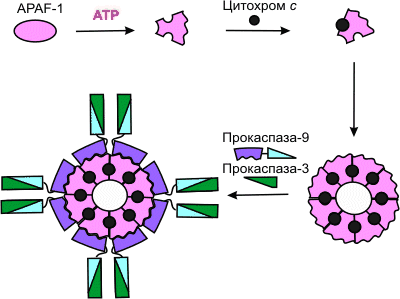
Флавопротеин AIF, будучи добавленным к изолированным ядрам из клеток HeLa, вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям печени крыс – высвобождение цитохрома с и каспазы- AIF является митохондриальным эффектором ПКС у животных, действующим независимо от каспаз .
Кроме рассмотренных компонентов, при нарушении наружной мембраны митохондрий из межмембранного объема выделяется термолабильный фактор, вызывающий необратимое превращение ксантиндегидрогеназы в ксантиноксидазу . Ксантиндегидрогеназа катализирует зависимое от NAD+ окисление ксантина до гипоксантина и последующее окисление гипоксантина до мочевой кислоты. Ксантиноксидаза катализирует те же реакции, но не с NAD+, а с О2 в качестве акцептора электронов. При этом образуются О2A, Н2О2, а из них – и другие активные формы кислорода (АФК), которые разрушают митохондрии и являются мощными индукторами апоптоза. Механизмы образования АФК, конечно, не ограничиваются ксантиноксидазной реакцией. Главным источником АФК в клетках являются митохондрии. Резкое увеличение АФК происходит при возрастании мембранного потенциала в митохондриях, когда снижено потребление ATP и скорость дыхания лимитируется ADP . Цитоплазматическая мембрана макрофагов и нейтрофилов содержит О2A – генерирующую NADPH-оксидазу.
В зависимости от пути, по которому осуществляется активация каспаз, различают разные типы клеток [82]. Клетки типа I (в частности, линия лимфобластоидных В-клеток SKW и T-клетки линии Н9) подвергаются ПКС по пути, зависимому от апоптозных рецепторов плазматической мембраны без участия митохондриальных белков. Клетки типа II (например, линии Т-клеток Jurkat и СЕМ) погибают по пути апоптоза, зависимому от митохондриального цитохрома с. ПКС, вызванная химиотерапевтическими соединениями, УФ- или і-облучением, по-видимому, напрямую связана с апоптозной функцией митохондрий.
Некоторые клетки, например, клетки эмбриональной нервной системы, включают механизмы апоптоза, если они испытывают дефицит апоптозподавляющих сигналов (называемых также факторами выживания) от других клеток. Физиологический смысл процесса – в элиминации избыточных нервных клеток, конкурирующих за ограниченный фонд факторов выживания. Эпителиальные клетки при отделении от внеклеточного матрикса, вырабатывающего факторы выживания, тоже обречены на ПКС. Факторы выживания связываются соответствующими цитоплазматическими рецепторами, активируя синтез подавляющих апоптоз агентов и блокируя стимуляторы апоптоза . Некоторые вещества (например, стероидные гормоны) оказывают дифференцированный эффект на различные типы клеток – предотвращают апоптоз одних типов клеток и индуцируют его у других [2].((((Так, при наличии во внеклеточном матриксе факторов роста PDGF (platelet-derived growth factor – тромбоцитарный фактор роста) или NGF (nerve growth factor – фактор роста нервов) и цитокина интерлейкина-3 (IL-3) проапоптозный белок Bad не активен .Факторы роста, связавшись со своим рецептором на плазматической мембране, вызывают активацию цитозольной протеинкиназы В, и катализирующей фосфорилирование Bad по Ser-136. IL-3 тоже связывается со своим рецептором на плазматической мембране и активирует митохондриальную cAMP-зависимую протеинкиназу А , катализирующую фосфорилирование Bad по Ser-112. Будучи фосфорилированным по обоим остаткам серина, Bad образует комплекс с белком 14-3-3, располагающийся в цитоплазме. Дефицит факторов роста и IL-3 воспринимается клеткой как сигнал к апоптозу: происходит дефосфорилирование Bad, его внедрение в наружную мембрану митохондрий, выход цитохрома с из митохондрий и последующая активация каспазы-9 через APAF-1-зависимый механизм. )))))