Трисомии и причины их возникновения
Таблица2. Основные врождённые пороки внутренних органов при синдроме Дауна (по Г. И. Лазюку с дополнениями)
|
Пораженная система и порок | Частота % общего числа больных |
|
Сердечно-сосудистая система | 53,2 |
|
Дефект межжелудочковой перегородки | 31,4 |
|
Дефект межпредсердной перегородки | 24,3 |
|
Открытый атриовентрикулярный канал | 9 |
|
Аномалии крупных сосудов | 23,1 |
|
Органы пищеварения | 15,3 |
|
Атрезия или стеноз двенадцатиперстной кишки | 6,6 |
|
Атрезия пищевода | 0,9 |
|
Атрезия прямой кишки и ануса | 1,1 |
|
Мегаколон | 1,1 |
|
Мочевая система (гипоплазия почек, гидроуретер, гидронефроз) | 5,9 |
Врождённые пороки внутренних органов, сниженная приспособленность детей с синдромом Дауна часто приводят к летальному исходу в первые 5 лет.
Следствием изменённою иммунитета и недостаточности репарационных систем (для повреждённой ДНК) являются лейкозы, часто встречающиеся у больных с синдромом Дауна. [8, 11]
Дифференциальная диагностика проводится с врождённым гипотиреозом, другими формами хромосомных аномалий. Цитогенетическое исследование у детей показано и при подозрении на синдром Дауна, и при клинически установленном диагнозе, поскольку цитогенетическая характеристика пациента необходима для прогноза здоровья будущих детей у родителей и их родственников. [14]
Этические проблемы при синдроме Дауна многоплановы. Несмотря на повышение риска рождения ребёнка с синдромом Дауна и другими хромосомными синдромами, врач должен избегать прямых рекомендаций по планированию беременности у женщин старшей возрастной группы, так как возрастной риск остаётся достаточно низким, особенно с учётом возможностей пре- натальной диагностики.
Неудовлетворённость у пациентов часто вызывает форма сообщения о синдроме Дауна у ребёнка. Поставить диагноз синдрома Дауна по фенотипическим признакам обычно можно немедленно после родо- разрешения. Врач, пытающийся отказаться от установления диагноза до исследования кариотипа, может потерять уважение родственников ребёнка. Важно сообщить родителям по крайней мере о ваших подозрениях как можно скорее после родоразрешения. Нецелесообразно полностью информировать родителей ребёнка с синдромом Дауна немедленно после родоразрешения. Нужно дать достаточно сведений, чтобы ответить на их немедленные вопросы и поддерживать их до того дня, когда станет возможно более детальное обсуждение. Немедленная информация должна включать объяснение этиологии синдрома для исключения взаимных обвинений супругов и описание исследований и процедур, необходимых для того, чтобы полностью оценить здоровье ребёнка. [1, 14]
Полное обсуждение диагноза нужно провести, как только родители, по крайней мере частично, оправятся от стресса родоразрешения, обычно в пределах 1-х суток. К этому времени у них возникает комплекс вопросов, на которые необходимо отвечать точно и определённо. На эту встречу приглашают обоих родителей. В этот период ещё слишком рано нагружать родителей всей информацией о заболевании, так как эти новые и сложные понятия требуют времени для восприятия. [1,8]
Не пытайтесь делать прогнозы. Бесполезно пробовать с точностью предвидеть будущее любого ребёнка. Древние мифы типа «по крайней мере он будет всегда любить и наслаждаться музыкой» непростительны. Важно отметить, что способности каждого ребёнка развиваются индивидуально. [1, 2, 3]
Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врождённые пороки сердца устраняют оперативно. Постоянно проводится общеукрепляющее лечение. Питание должно быть полноценным. Необходимы внимательный уход за больным ребёнком, защита от действия вредных факторов окружающей среды (простуда, инфекции). Многие больные с три- сомией 21 теперь способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семьи. [1]
ГЛАВА 3. СИНДРОМ ЭДВАРДСА – ТРИСОМИЯ 18
При цитогенетическом исследовании обычно обнаруживают регулярную трисомию 18. Как и при синдроме Дауна, выявляется связь между частотой трисомии 18 и возрастом матери. В большинстве случаев дополнительная хромосома имеет материнское происхождение. Около 10 % трисомии 18 обусловлены мозаицизмом или несбалансированными перестройками, чаще робертсоновскими транслокациями. [2]
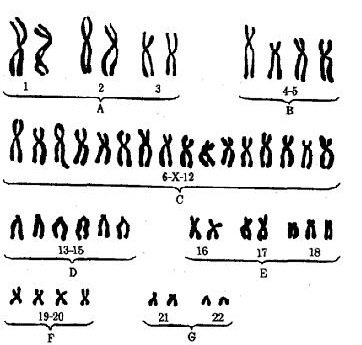
Рис. 7 Кариотип Трисомия 18
Клинических различий между цитогенетически различающимися формами трисомии нет. [1]
Частота синдрома Эдвардса составляет 1:5000—1:7000 новорождённых. Соотношение мальчиков и девочек равно 1:3. Причины преобладания больных девочек пока неясны.
При синдроме Эдвардса отмечается выраженная задержка пренатального развития при полной продолжительности беременности (роды в срок). На рис. 8-9 представлены пороки развития, характерные для синдрома Эдвардса. В первую очередь это множественные врождённые пороки развития лицевой части черепа, сердца, костной системы, половых органов. [1,2]
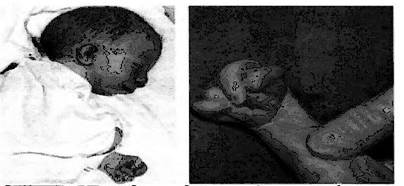
Рис. 8 Новорожденный с Рис. 9 Характерное для синдромом Эдвардса. синдрома Эдвардса Выступающий затылок; положение пальцев микрогения; флексорное (возраст ребенка 2 мес.) положение кисти
Череп долихоцефалической формы; нижняя челюсть и отверстие рта маленькие; глазные щели узкие и короткие; ушные раковины деформированные и низко расположенные. Из других внешних признаков отмечаются флексорное положение кистей, аномально развитая стопа (пятка выступает, сводно провисает), I палец стоп короче II. Спинномозговая грыжа и расщелина губы встречаются редко (5% случаев синдрома Эдвардса).
Многообразная симптоматика синдрома Эдвардса у каждого больного проявляется лишь частично. Частота отдельных врожденных пороков приведена в табл. 3. [1]
Таблица3. Основные врождённые пороки при синдроме Эдвардса (по Г. И. Лазюку)
|
Пораженная система и порок (признак) | Относительная частота, % |
|
Мозговой череп и лицо | 100,0 |
|
микрогения | 96,6 |
|
низко расположенные и(или) деформированные ушные раковины | 95,6 |
|
долихоцефалия | 89,8 |
|
высокое нёбо | 78,1 |
|
расщелина нёба | 15,5 |
|
микростомия | 71,3 |
|
Опорно-двигательный аппарат | 98,1 |
|
флексорное положение кистей | 91,4 |
|
дистальное расположение I пальца кисти | 28,6 |
|
гипоплазия и аплазия I пальца кисти | 13,6 |
|
короткий и широкий I палец стопы | 79,6 |
|
стопа-качалка | 76,2 |
|
кожная синдактилия стоп | 49,5 |
|
косолапость | 34,9 |
|
короткая грудина | 76,2 |
|
ЦНС | 20,4 |
|
гипоплазия и аплазия мозолистого тела | 8,2 |
|
гипоплазия мозжечка | 6,8 |
|
Глаза (микрофтальмия) | 13,6 |
|
Сердечно-сосудистая система | 90,8 |
|
дефекты межжелудочковой перегородки | 77,2 |
|
в том числе входящие в комбинированные пороки | 65,4 |
|
дефекты межпредсердной перегородки | 25,2 |
|
в том числе входящие в комбинированные пороки | 23,8 |
|
аплазия одной створки клапана лёгочной артерии | 18,4 |
|
аплазия одной створки клапана аорты | 15,5 |
|
Органы пищеварения | 54,9 |
|
дивертикул Меккеля | 30,6 |
|
незавершённый поворот кишечника | 16,5 |
|
атрезия пищевода | 9,7 |
|
атрезия желчного пузыря и жёлчных ходов | 6,8 |
|
эктопия ткани поджелудочной железы | 6.8 |
|
Мочевая система | 56.9 |
|
сращение почек | 27,2 |
|
удвоение почек и мочеточника | 14.6 |
|
кисты почек | 12,6 |
|
гидро- и мегалоуретер | 9,7 |
|
Половые органы | 43,5 |
|
крипторхизм | 28,6 |
|
гипоспадия | 9,7 |
|
гипертрофия клитора | 16,6 |