Трисомии и причины их возникновения
Как видно из табл. 3, наиболее значимыми в диагностике синдрома Эдвардса являются изменения мозгового черепа и лица, опорно-двигательного аппарата, пороки развития сердечно-сосудистой системы. [2,3]
Дети с синдромом Эдвардса умирают в раннем возрасте (90% — до 1 года) от осложнений, обусловленных врождёнными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность). Клиническая и даже патологоанатомическая дифференциальная диагностика синдрома Эдвардса сложна. Во всех случаях показано цитогенетическое ис- следование. [1] Диагностика синдрома Эдвардса особенно затруднена во время беременности, несмотря на наличие такого эффективного метода диагностики аномалий развития плода, как УЗИ. Косвенными признаками по данным УЗИ, указывающими на синдром Эдвардса у плода, могут стать малая величина плаценты, недоразвитие или отсутствие одной из пупочных артерий в пупочном канатике. На ранних сроках УЗИ не позволяет обнаружить в случае синдрома Эдвардса каких-либо грубых аномалий развития. Из-за данной совокупности диагностических трудностей вопроса о своевременном прерывании беременности обычно не возникает, и женщины до конца вынашивают таких детей. Какого-либо метода лечения синдрома Эдвардса не существует. [12]
ГЛАВА 4. СИНДРОМ ПАТАУ – ТРИСОМИЯ 13
Синдром Патау выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования, проведённого у детей с врождёнными пороками развития. Частота синдрома Патау среди новорождённых равна 1:5000—1:7000. Цигогенетические варианты этого синдрома следующие. Простая полная трисомия 13 как следствие нерасхождения хромосом в мейозе у одного из родителей (главным образом у матери) встречается у 80—85% больных. Остальные случаи обусловлены в основном передачей дополнительной хромосомы (точнее, её длинного плеча) в робертсоновских транслокациях типа D/13 и G/13. Обнаружены и другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации), но они встречаются крайне редко. Клиническая и патологоанатомическая картина простых трисомных форм и транслокационных не различается. [1, 2]
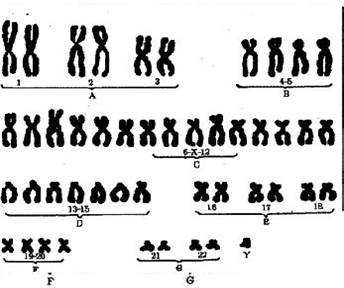
Рис. 10 Кариотип Трисомия 13
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25—30% ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок гестации 38,3 нед). Характерное осложнение беременности при вынашивании плода с синдромом Патау — многоводие: оно встречается почти в 50% случаев синдрома Патау. [1]
Для синдрома Патау характерны множественные врожденные пороки развития головного мозга и лица (рис. 11).
Это патогенетически единая группа ранних (и, следовательно, тяжёлых) нарушений формирования головного мозга, глазных яблок, мозговой и лицевой частей черепа. Окружность черепа обычно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные.
Типичный признак синдрома Патау — расщелины верхней губы и нёба (обычно двусторонние). Всегда обнаруживаются пороки нескольких внутренних органов в разной комбинации: дефекты перегородок сердца, незавершенный поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы. Как правило, наблюдаются полидактилия (чаще двусторонняя и на руках) и флексорное положение кистей. Частота разных симптомов у детей с синдромом Патау представлена в табл. 4. [1, 7]
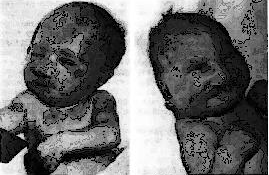
Рис. 11 Новорождённый с синдромом Патау. Тригоноцефалия (б); двусторонняя расщелина верхней губы и нёба (б); узкие глазные щели (б); низко расположенные (б) и деформированные (а) ушные раковины; микрогения (а); флексорное положение кистей
Клиническая диагностика синдрома Патау основывается на сочетании характерных пороков развития. При подозрении на синдром Патау показано УЗИ всех внутренних органов. [1]
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95% — до I года). Однако некоторые больные живут несколько лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных с синдромом Патау до 5лет (около 15% детей) и даже до 10 лет (2—3% детей).
Таблица4. Основные врождённые пороки при синдроме Патау (по Г. И. Лазюку)
|
Пораженная система и порок | Относительная частота, % |
|
Лицо и мозговой череп | 96,5 |
|
низко расположенные и(или) деформированные ушные раковины | 80,7 |
|
расщелина верхней губы и нёба | 68,7 |
|
в том числе только нёба | 10,0 |
|
микрогения | 32,8 |
|
дефект скальпа | 30,8 |
|
Опорно-двигательный аппарат | 92,6 |
|
полидактилия кистей | 49,0 |
|
полидактилия стоп | 35,7 |
|
флексорное положение кистей | 44,4 |
|
стопа-качалка | 30,3 |
|
ЦНС | 83,3 |
|
аринэнцефалия | 63,4 |
|
в том числе голопрозэнцефалия | 14,5 |
|
микроцефалия | 58,7 |
|
аплазия и гипоплазия мозолистого тела | 19,3 |
|
гипоплазия мозжечка | 18,6 |
|
в том числе гипоплазия и аплазия червя | 11,7 |
|
аплазия и гипоплазия зрительных нервов и трактов | 17,2 |
|
Глазное яблоко | 77,1 |
|
микрофтальмия | 70,5 |
|
колобома радужки | 35,3 |
|
помутнение хрусталика | 25,9 |
|
анофтальмия | 7,5 |
|
Сердечно-сосудистая система | 79,4 |
|
дефект межжелудочковой перегородки | 49,3 |
|
в том числе компонент комбинированного порока | 44,8 |