Цитогенетическая и молекулярно-цитогенетическая характеристика микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи
Продолжительность жизни больных может достигать 60 лет и более.
Согласно данным литературы, патогенез синдрома Прадера-Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза.
Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков [5].
Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке.
Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера-Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может оказать роковое влияние на развитие злокачественных новообразований у лиц с синдромом Прадера-Вилли [5].
Терапия синдрома Прадера-Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины).
СПВ должен быть заподозрен у детей до 3-летнего возраста при наличии не менее 5 баллов, а у детей старше 3 лет — 8 баллов, при условии присутствия 4 и более больших признаков.
Несмотря на высокую диагностическую чувствительность приведенной шкалы (около 90%), во всех случаях для подтверждения диагноза требуются кариотипирование и молекулярно-генетические исследования 15-й пары хромосом. Специфичность этих исследований достигает 100% . Кроме того, от выявленного типа генетических нарушений зависит генетический прогноз потомства.
Дети, страдающие СПВ, должны постоянно находиться под наблюдением педиатра, невролога, психотерапевта, эндокринолога и офтальмолога.
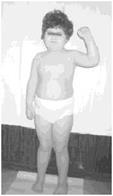
Рис. 4. Девочка с синдромом Прадера-Вилли
2.2.2 Клинические проявления синдрома Ангельмана
В 1965 году доктор Гарри Ангельман, английский психиатр, впервые описал трех детей с характеристиками, в настоящее время известными как синдром Ангельмана. Он отметил, что все эти дети имели неуклюжесть, трясущуюся походку, отсутствие речи, излишне смешливы и впадали в припадки. Другие похожие случаи уже были описаны учеными, но эти случаи были особенными, и многие психиатры подтвердили их исключительность. Первые отчеты пришли из Северной Америки в начале 1980-х гг.
Синдром Ангельмана — генетическая аномалия. Для него характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки.
Частота встречаемости, по разным данным, — 1: 10 000-20 000 живорожденных младенцев. (Однако, согласно данным Центра развития человека и отклонений в развитии (университет Вашингтона, США), можно предполагать, что доля людей с синдромом Ангельмана в действительности намного больше статистической.)
Синдром Ангельмана обычно не распознается при рождении или в раннем детстве, пока не проявят себя проблемы в развитии, которые не специфичны к этому времени. Родители могут заподозрить диагноз после прочтения о синдроме Ангельмана или после встречи ребенка с такими же признаками. Наиболее распространенный возраст для диагностики – между тремя и семью годами, когда отличительные черты поведения становятся более очевидными.
Для синдрома Ангельмана характерны:
- В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес;
- задержка в развитии навыков общей моторики (умение сидеть, ходить);
- задержка речевого развития, неразвитая речь (у всех детей);
- дети больше понимают, чем могут сказать или выразить;
- дефицит внимания и гиперактивность;
- сложности с обучением;
- эпилепсия (80% случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана происходит вторичная (симптоматическая) общая эпилепсия;
- необычные движения (мелкий тремор, хаотические движения конечностей);
- частый смех без повода;
- ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками;
- размер головы меньше среднего, нередко с уплощением затылка;
- иногда особые черты лица — широкий рот, зубы с промежутками между ними, выдающийся вперед подбородок, высунутый наружу язык);
- нарушения сна;
- страбизм (косоглазие) в 40 % случаев;
- сколиоз (искривление позвоночника) в 10 % случаев;
- повышенная чувствительность к высокой температуре;
- бывают сильно увлечены играми с водой [7].
Диагностика: синдром диагностируется путем генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи.
Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15q11-q13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE3A.
Существует небольшая группа людей, у которых результаты всех вышеописанных анализов в норме, однако наблюдаются все внешние проявления синдрома Ангельмана. Наука пока не дает ответ на вопрос, как это возможно.
Лечение: синдром Ангельмана является врожденной генетической аномалией и, следовательно, не может быть излечен.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом.
В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии).
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0.3 мг мелатонина за 30 минут-1 час перед сном улучшает сон пациентов с синдромом Ангельмана. А нарушения стула регулируются назначением легких слабительных. Приступы лечатся так же, как эпилепсия.
Дети с синдромом Ангельмана часто испытывают больше одного типа приступов. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложность в общении с другими людьми. Врачи США показывают, что для аутичных детей внутривенные инъекции гормона секретина успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана [2, 7].