Анализ сополимеризации индена с малеиновым ангидридом
1. Скорость распада пероксида бензоила сильно зависит от растворителя, в котором протекает реакция.
2. Кинетический порядок реакции распада также зависит от растворителя.
3. При уменьшении концентрации пероксида удельная скорость распада (скорость, отнесённая к начальной концентрации пероксида) во многих случаях уменьшается и для различных растворителей стремится к одинаковой величине.
4. Добавление некоторых веществ, в частности виниловых соединений, к растворителям, в которых протекает быстрый распад пероксида, приводит к снижению удельной скорости реакции до величины, наблюдаемой при распаде пероксида в разведенных растворах.
Таким образом, распад пероксида представляет цепную реакцию [6-8], причём длина цепей зависит от природы растворителя. Предложено [6] следующее выражение для скорости распада пероксида бензоила:
![]() , (2.4)
, (2.4)
где (ПБ)-концентрация пероксида бензоила, k1 –константа скорости первичного мономолекулярного распада, а член kц(ПБ)n характеризует скорость цепного распада пероксида.
При виниловой полимеризации скорость и эффективность инициирования определяется первичным распадом пероксида, поэтому важно знать константу скорости мономолекулярного распада пероксида kПБ. Данные распада пероксида бензоила в бензоле (начальная концентрация 0,00185 моль/л) при температуре 60-80ºС удовлетворяют уравнению [5]:
![]() (2.5)
(2.5)
В ароматических растворителях, например в толуоле, цепной распад пероксида бензоила при концентрациях пероксида не больше 0,2 моль/л невелик и приводит к образованию несимметричных дифенилов и значительных количеств бензойной кислоты (~50% от теории). Лёгкость присоединения фенильного радикала к бензольному кольцу с образованием нереакционного радикала позволяет понять малую величину цепного распада пероксида в ароматических растворителях. По-видимому, бензоатные радикалы также могут присоединяться к ароматическому кольцу, что приводит к образованию эфира бензойной кислоты. Присоединение бензоатного радикала к бензолу протекает медленнее, чем декарбоксилирование, так как выход эфира составляет лишь 5-7% [9].
Цепной распад пероксида может быть подавлен добавками ингибиторов. Особенно эффективны в этом отношении виниловые соединения. Поведение виниловых соединений по отношению к пероксиду бензоила во многих отношениях аналогично поведению ароматических соединений. Бензоатные радикалы, первоначально образующиеся при распаде пероксида бензоила, могут или присоединяться к двойной связи, давая начало полимерным цепям, или отщеплять молекулу диоксида углерода с образованием фенильного радикала, который также может присоединяться к двойной связи. Конкуренцию между реакцией декарбоксилирования и реакцией присоединения к двойной связи исследовали по выходу диоксида углерода при распаде пероксида бензоила в присутствии мономера. При увеличении концентрации мономера выход СО2 уменьшается, а при равных концентрациях зависит от природы мономера.
За инициированием следует стадия роста цепи [1, 2]. На этой стадии активный центр, находящийся на первом мономерном звене, атакует двойную связь следующей молекулы мономера. Эта атака приводит к присоединению второго мономерного звена и переносу активного центра с первого на второе мономерное звено в соответствии со следующей схемой:
![]()
Необходимо отметить, что этот активный центр вновь способен к атаке следующей молекулы мономера с дальнейшим переносом неспаренного электрона на конец растущей цепи. Такой процесс, включающий последовательность актов присоединения молекул мономера к активному центру, получил название роста цепи. Возможны различные типы присоединения молекул мономера к активному концу растущей цепи. Так, различают присоединение по типу «голова к хвосту», «хвост к хвосту», «голова к голове», «хвост к голове», при этом под «головой» и «хвостом» мономерного звена понимают группы –СНХ– и –СН2–. Как упоминалось выше, присоединение очередного мономерного звена к концу растущей цепи сопровождается переходом p-электронной пары на уровень s-электронов и выделением энергии ~ 20 ккал. Следовательно, необходимо небольшое количество энергии извне для разложения инициатора и образования свободных радикалов, а далее полимерные цепи начинают рост с выделением большого количества энергии.
Поскольку при разложении инициатора одновременно образуется большое количество свободных радикалов, каждый из которых инициирует и продолжает рост цепей, в системе в любой момент времени существует определённое количество растущих цепей. В зависимости от ряда факторов, таких как температура, время реакции, концентрация мономера и инициатора, существует некоторая статистическая вероятность сближения двух растущих цепей и их взаимного столкновения. Когда происходит такое взаимное проникновение растущих клубков, возможно осуществление двух реакций, приводящих к обрыву цепей:
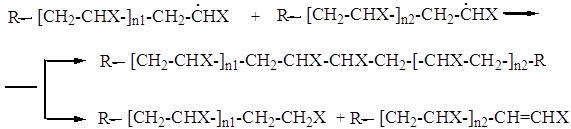
В первом случае две растущие цепи соединяются за счёт спаривания одиночных электронов активных центров, поэтому такой вид обрыва получил название рекомбинации. Во втором случае атом водорода отрывается от конца одной из растущих цепей, присоединяется к концу второй и образует стабильную связь с неспаренным электроном активного центра. Цепь, отдавшая атом водорода, стабилизируется за счёт образования концевой двойной связи. Такая реакция обрыва приводит к образованию двух полимерных молекул и носит название диспропорционирования.
Существует ещё один вид реакций ограничения длины растущих цепей, который происходит путём «передачи цепи» [2]. Эта реакция протекает обычно путём отрыва атома водорода или любого другого подвижного атома от молекулы инициатора, мономера, мёртвых полимерных цепей или любых молекул, присутствующих в реакционных системах, включая растворитель и примеси. Ее можно схематически представить следующим образом:
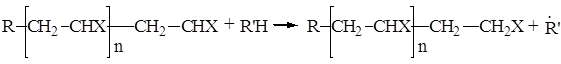
Растущая цепь при этом обрывается, но образуется новый радикал ![]() . Он теперь способен инициировать рост новой полимерной цепи. Таким образом, в ходе этой реакции одна живая, растущая цепь прекращает свой рост, тогда как новая начинает всё сначала.
. Он теперь способен инициировать рост новой полимерной цепи. Таким образом, в ходе этой реакции одна живая, растущая цепь прекращает свой рост, тогда как новая начинает всё сначала.
1.1.2 Полимеризация виниловых мономеров в присутствии комплексообразователей типа кислот Льюиса
Известно, что органические основания Льюиса (нитрилы, сложные эфиры, амиды и т. п.) способны образовывать достаточно прочные координационные комплексы с координационно-ненасыщенными соединениями непереходных металлов (чаще всего Zn, B, Al, Sn и др.), выступающих в таких случаях в качестве Льюисовых кислот. Непредельные органические соединения указанных выше классов, в частности, практически важные акриловые и метакриловые мономеры обычно полимеризуются по радикальному механизму. Образование их комплексов с кислотами Льюиса, естественно, приводит к изменению электронной структуры молекул мономеров и, следовательно, их реакционной способности. Поэтому кислоты Льюиса могут быть использованы в качестве модификаторов для целенаправленного изменения кинетических параметров отдельных элементарных стадий полимеризационных процессов.