Технология нуклеофильного замещения функциональных групп в органических соединениях
 (9)
(9)
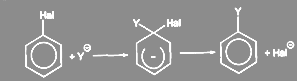
Существование таких отрицательно заряженных σ-комплексов было доказано экспериментально:
 (10)
(10)
Подобные интермедиаты представляют собой устойчивые соли, называемые солями Мейзенгеймера, со времени обнаружения их в 1902 г было выделено большое число таких солей, строение нескольких интермедиантов такого типа было подтверждено данными ЯМР и pентгеноструктурного анализа. Однако описанный механизм не является единственно возможным. С помощью меченого атома углерода было показано что в арилгалогенидах, не содержащих активирующих групп, замещающая группа становится не только к тому атому углерода, где был галоген, но в равной степени и к соседнему атому:
 (11)
(11)
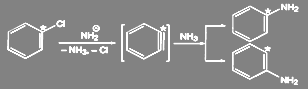
Идентичность соседних положений при отсутствии других заместителей в ядре объясняется тем, что реакция идет по механизму отщепления-присоединения (кинезамещения) через стадию образования 1,2-дегидродензола:
 (12)
(12)
Промежуточное образование дегидробензола было доказано как физико-химическими, так и чисто химическими методами. Так, при действии амальгамы лития на l-фтор-2-бромбензол в присутствии диенофилов промежуточно образующийся 1 ,2-дегидробензол вступает с ними в реакцию Дильса-Альдера:
1.2 Основные факторы влияющие на ход процесса нуклеофильного замещения
Условия проведения и ход реакций нуклеофильной замены галогена зависит от многих факторов. При выборе оптимальных условий проведения процесса необходимо учитывать особенности химического строения субстрата и нуклеофильного реагента, полярность среды, природу уходящего галогена.
Относительно связи строения субстрата и его реакционной способности нужно отметить следующее. Скорости SN1 реакций алкильных производных возрастают в ряду: первичный алкил, вторичный, третичный. Так, константы скоростей реакций гидролиза алкилбромидов при 50 ºС для R-С2Н5; (СН3)2СН-; (СН3)3С- относятся соответственно как 1 : 11,6 : 1,2·106. Пространственные препятствия в этом случае не имеют большого значения. Более того, увеличение объема заместителей дестабилизирует исходное состояние в большей степени, чем переходное, что должно приводить к увеличению скорости диссоциации. Особенности строения молекулы субстрата, приводящие к стабилизации образующегося карбкатионa, должны приводить к ускорению реакции SN1 замещения. Это достигается, в частности, при наличии в α-положении к реакционному центру фенильных или аллильных заместителей, а также атомов, имеющих неподелённую пару электронов.
При этом по силе активации один α-фенильный радикал соответствует примерно двум алкильным заместителям. [3]
Что касается влияния строения субстрата на скорость SN2 замещения то порядок изменения реакционной способности при переходе первичного к третичному радикалу прямо противоположен наблюдаемому при SN1 замещении. Первичные галогенпроизводные реагируют очень гладко, вторичные - значительно хуже, а третичные часто не реагируют вооще. Пространственные эффекты играют в SN2 замещении важную роль, и низкая скорость для третичных галогенидов объясняется, в частности пространственными препятствиями для атаки нуклеофилом.
Таким образом, при переходе от первичного алкилгалогенида к тритичному механизм реакции может измениться от бимолекулярного до мономолекулярного. Переход от одного механизма к другому не является резким и зависит от ряда конкретных условий. Принципиально возможно протекание реакции по двум механизмам одновременно.
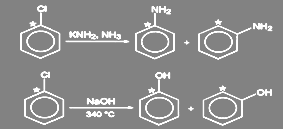
В ароматических галогенидах, как уже отмечалось, замещение практически всегда происходит по бимолекулярному механизму. Исключением является разложение солей диазония. Влияние других заместителей в ароматическом кольце на легкость замещения галогена изучалось очень широко. Наличие электроноакцепторных заместителей в орто-, пара- положениях существенно облегчает реакцию SN 2 замещения, электродонорных - затрудняет ее.
Сильное ускорение процесса замещения галогена под влиянием орто- и пара-расположенных нитрогрупп хорошо известно. Так, 2,4,6-тринитрогалогенбензолы очень легко реагируют с водой, спиртами, аммиаком, первичными и вторичными аминами, образуя пикриновую кислоту, с эфиры или амиды. Динитрогалогенбензолы реагируют с подобными реагентами медленнее, а мононитро - значительно медленнее. Так, пикрилхлорид гидролизуется так же легко, как хлорангидрид кислоты, замена галогена в о- и п-хлорнитробензоле проходит в щелочном растворе при 130-150 º С, а хлорбензол гидролизуется до фенола лишь при температуре: 350-400 º С и давлении выше 30 МПа под действием 5% раствора щелочи.
В орто- и пара-замещенных хлорбензолах легкость замещения хлора на гидроксил определяется рядом: N02 > > SO3H > СООН. При этом активация за счет NО2-группы на несколько порядков выше активации за счет SO3H и СООН-групп. [10]
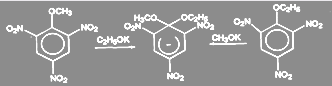
При взаимодействии замещенных галогенбензолов с метилатом натрия активирующее действие групп при одинаковом их размещении относительно галогена изменяется в соответствии с рядами:
![]() (14)
(14)
Отмечено значительное увеличение подвижности галогена в ароматическом ядре при наличии в орто- и пара- положении к нему NH3 –группы (т. е. аминогруппы в кислой среде).
Заместители первого рода значительно снижают подвижность галогена и в ряде случаев переводят механизм SNАг в кинезамещение через дегидробензол. При переходе к пиридину и хинолину нуклеофильная подвижность галогена повышается. В этом смысле пиридин и хинолин можно рассматривать как аналоги нитробензола. 4-Галогенпиридины активнее 2-замещенных; 3-галогензамещенные еще менее активны и в этом смысле похожи на фенилгалогениды.
При переходе к диазинам нуклеофильная подвижность атома галоген увеличивается. Высокой реакционной способностью выделяются 2- и 4-галогенпиримядины. 2-Хлорпиримидин реагирует с н-бутиламином уже при комнатной температуре, а 4-хлорпиримидин нельзя выделить в индивидуальном состоянии из-за легкого отщепления хлора. 2-Хлорпиразин и 3-хлорпиридазин также значительно активнее 2-хлорпиридина в реакциях нуклеофильного замещения.
В ряду пятичленных гетероциклов реакции нуклеофильного замещения изучены еще недостаточно. Галогензамещенные фураны и тиофены относительно инертны в реакциях нуклеофильного замещения, хотя их реакционная способность выше, чем у соответствующих галогенарилов. Введение сильных электроноакцепторных заместителей увеличивает подвижность галогена.
Пространственные факторы при нуклеофильном замещении в ароматическом ряду не являются определяющими, так как атака направлена сбоку к плоскости ароматического ядра.
В зависимости от природы галогена порядок реакционной способности алкилгалогенидов в реакциях нуклеофильного замещения оказывается следующим: RI> RВr> RСI> RF. Иное положение наблюдается для является не переходным состоянием, а промежуточным соединением. Величина положительного заряда у реакционного центра зависит не только от количества, расположения и природы других заместителей в ядре, но и от природы замещаемого галогена. Поэтому в активированных ароматических системах атомы галогена могут быть замещены с возрастающей легкостью в ряду I < Вr < СI < F.[13]