Технология нуклеофильного замещения функциональных групп в органических соединениях
 (37)
(37)
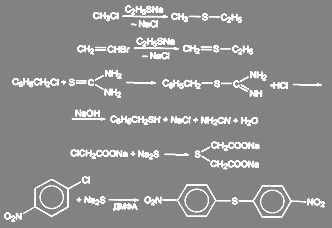
Симметричные и смешанные сульфиды могут быть получены и в условиях МФК. Так, например, с высокими выходами образуются несимметричные тиоэфиры из первичных и вторичных алкилбромидов при их взаимодействии с меркаптанами и тиофенолами в присутствии водных растворов щелочи. В качестве катализаторов межфазного переноса использовались ониевые соли, краун-эфиры и криптаты. детально изученный механизм реакции тиофенола с н-бромоктаном в щелочной двухфазной системе подтверждает SN2 характер замещения:
![]() (38)
(38)
1.4 Замена атома галогена на группы -NН2, -NНR, -NR2
Алкил- и арилгалогениды могут взаимодействовать с аммиаком и аминами. Эти реакции можно рассматривать также и как частный случай N-алкилирования.
Если нагревать алкилгалогенид со спиртовым (реже водным или водноспиртовым) раствором аммиака, первичного или вторичного амина под давлением и при высокой температуре (в автоклаве), то образуется смесь солеи соответствуюших аминов (первичных, вторичных, третичных), а так же четвертичных солей аммония (реакция Гофмана):
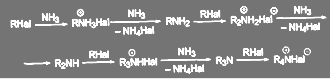 (39)
(39)
Вместо аммиака можно использовать первичные, вторичные или третичньие амины (реакция Меншучкина) и получать смешанные аминосоединения:
![]() (40)
(40)
Третичные алкилгалогениды в этих реакциях обычно не применяют, так как в условиях реакции идет элиминирование с образованием алкенов.
Выход первичного амина можно повысить, применяя большой избыток аммиака и добавляя карбонат или хлорид аммония. Однако даже в этом случае образуется смесь соединений, которые приходится разделять.
Поскольку для синтеза биологически активных веществ особенно важно иметь индивидуальные и чистые вещества строго определённой структуры, особое значение приобретают те методы и приёмы, которые позволяют достичь такого результата.
Один из возможных путей синтеза вторичных аминов без примеси третичных ясен из схемы:
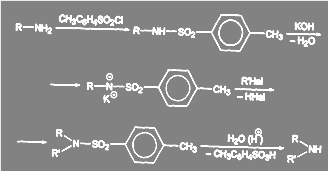 (41)
(41)
Другим способом получения вторичных аминов является синтез через азометины:
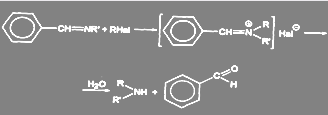 (42)
(42)
Первичные амины можно получить гидролизом и гидразинолизом алкилфталимида (синтез Габриэля):
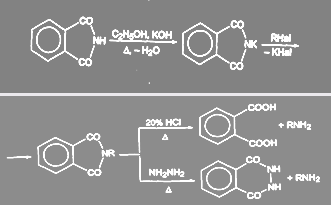
Преимущество гидразинолиза перед гидролизом состоит в том, что гидролиз приходится проводить при высоких температурах под давлением, в то время как взаимодействие с гидразином идет при нормальном давлении.
Допустимые выходы первичных аминов дают лишь α-галогенкарбоновые кислоты при действии большого избытка аммиака. Таким образом получают:
 (45)
(45)
Реакцию ведут, нагревая при 40—50 °С соответствуюшую галогензамещенную кислоту с концентрированным водным раствором аммиака и карбоната аммония. Выходы α-аминокислот — 60—70%.
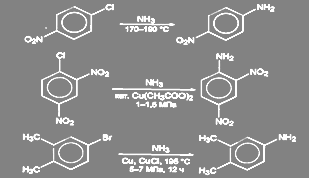
В неактивированных галогенпроизводных ароматического ряда галоген может быть замещен на аминогруцпу действием раствора аммиака при высокой температуре и давлении в присутствии катализатора (Сu2O, СuSO4, и т.д.). Другой способ состоит в действии амид-иона в жидком аммиаке. Реакция в этом случае идёт через образование дегидробензола.[2]
Условия проведения реакции в активированных соединениях зависят от степени активации галогена:
 (46)
(46)
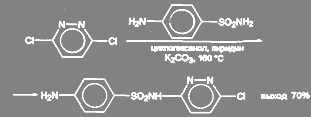
3-хлор-6-(п-аминбензолсульфаниламидо)-пиридазин получают из 3,6-дихлорпиридазина. Реакцию ведут в среде циклогексанола с добавкой пиридина и значительного количества поташа (2 моля на 1 моль) при температуре 135 – 160 ºС: T5r
 (47)
(47)
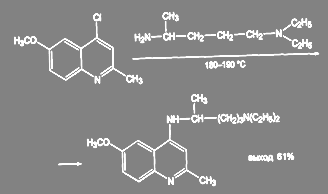
Получение 2-метил-4-(α-метил-δ-диэтиламинобутиламино)-6-метоксихинолина в производстве трихомонацида ведут в более жёстких условиях, в этом случае требуется двойной избыток диэтиламино-4-аминопентана и температура 185 – 190 ºС:
 (48)
(48)
При получении антидеприсанта азафена аминирование ведут дигидрохлоридом N-метилпиперазина:
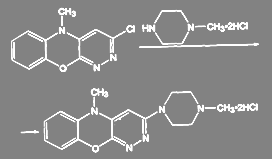 (49)
(49)
Получение амидов из хлорангедридов кислот идёт в мягких условиях. Так, получение диакарба проводят при температуре 3-4 ºС действием охлаждённого раствора аммиака на соответствующий сульфохлорид:
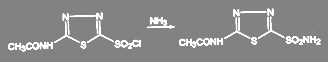 (50)
(50)
Аналогичные реакции имеют место при получении метилуретана:
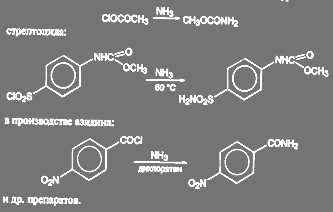 (51)
(51)
1.5 Замена атома галогена на - СN и - SO3Na
Замена галогена на СN-группу является относительно простым способом удлинения углеродной цепи. Этот способ, однако, неприменим для пространственно затрудненных субстратов. Первичные галогениды, а также бензил- и аллилгалогениды дают хорошие выходы нитрилов. В случае вторичных алкилгалогенидов выходы средние. С третичными галогенидами реакция не идет, так как в этих условиях проходит реакция не замещения, а элиминирования, Для проведения синтеза пригодны многие растворители, но наилучшие результаты достигаются в диметилсульфоксиде (ДМСО).[6]
Реакционная способность алкилгалогенидов увеличивается в ряду CI< Вr <I , что позволяет проводить реакцию избирательно:
![]() (52)
(52)
Поскольку цианид-ион является амбидентнsм ионом, реакция может идти по двум направлениям — с образованием нитрилов и изонитрилов:
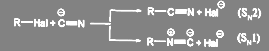 (53)
(53)
В случае первичных алифатических галогенидов и бензилгалогенидов в спиртах и водно-спиртовых смесях нежелательная примесь изонитрилов не образуется или образуется в очень малых количествах (легко обнаруживается по крайне неприятному запаху).
В случае ароматических галогензамещенных реакцию следует вести в апротонных растворителях. Галогенбезолы могут также превращаться в соответствующие фенилцианиды при нагревании до 200 °С с цианидом меди (1) в растворе пиридина:
![]() (54)
(54)
Реакционноспособные алкилгалогениды переводят в нитрилы кипячением галогенида и цианида натрия в сухом ацетоне с добавлением небольшого количества (0,05 моля на 1 моль субстрата) иодида натрия. Для инетных галогенидов в качестве растворителя используют 70—90% спирт или триэтиленгликоль. Правильный выбор растворителя во многом определяет успех реакции. Так, например, алифатические нитрилы можно получать с высоким выходом в ДМСО или ДМФА. Наибольшие примеси изонитрила можно гидролизовать в кислой среде и таким образом отделить от основного продукта, так как нитрилы гидролизуются в значительно более жестких условиях.[17]