Реакции нуклеофильного замещения
Изменение реакционной способности нуклеофила может также быть следствием того, что в зависимости от растворителя нуклеофил может находиться в виде ионов или ионных пар. Связывание нуклеофила в ионную пару уменьшает его реакционную способность.
Так как образование ионных пар тем вероятнее, чем меньше размеры катиона и аниона, реакционная способность уменьшается в наибольшей степени именно для солей с малыми анионами. Ниже указана реакционная способность ионов и ионных пар в реакции C4HgOBs + Х- в ацетоне при 250С:
| M+X- | kотнсумм | kд * 104 | kотнсв.ион |
|
(C4H9)4N+Cl- | 11,3 | 22,8 | 18 |
|
Li+Cl- | 0,16 | 0,027 | 18 |
|
(C4H9)4N+Br- | 3,0 | 32,9 | 4 |
|
Li+Br- | 0,92 | 5,22 | 4 |
|
(C4H9)4N+I- | 0,6 | 64,8 | 1 |
|
Li+I- | 1,0 | 69 | 1 |
|
kд – константа диссоциации ионных пар | |||
Из этих данных видно, что для галогенидов лития, практически полностью находящихся в виде ионных пар, наиболее реакционноспособным оказывается иодид. В то же время образование ионных пар в случае тетрабутиламмонийгалогенидов является значительно менее вероятным. Измерение констант равновесия ионы – ионные пары позволило рассчитать относительные реакционные способности свободных галоген-анионов. Как и следовало ожидать, они изменяются в том же порядке, что и в других апротонных растворителях.
В последние годы использование краун-эфиров, которые образуют комплексы с катионами и тем самым разрушают ионные пары, позволило более широко изучить нуклеофильные реакции в апротонных растворителях.
Таким образом, очевидно, что поляризуемость нуклеофила не играет существенной роли. Успех уравнения Эдвардса, которое мы рассматривали, вероятно, связан с тем, что поляризуемость и сольватация нуклеофила изменяются в противоположных направлениях: чем больше объем аниона, тем выше его поляризуемость и тем меньше его сольватация. Исследование реакций замещения в газовой фазе также продемонстрировало определяющую роль сольватации для реакционной способности нуклеофилов. Было показано, что в реакциях анион - молекула рост основности нуклеофила приводит к увеличению его реакционной способности, а поляризуемость не играет существенной роли. Из галоген-анионов наиболее реакционноспособным является фторид-ион. В то же время различие между реакционной способностью нуклеофилов в газовой фазе значительно меньше, чем в растворе. Это связано с тем, что реакции в газовой фазе идут с очень большими скоростями. Таким образом, еще раз подтверждается вывод о том, что очень большой вклад в энергию активации реакций бимолекулярного замещения вносит энергия десольватации реагентов.
КОНКУРЕНЦИЯ МОНО- И БИМОЛЕКУЛЯРНОГО ЗАМЕЩЕНИЯ.
В предыдущих разделах мы рассмотрели основные факторы, влияющие на скорость реакций бимолекулярного замещения. Рассмотрим теперь, как можно изменять механизм реакции замещения, т. е. варьируя строение субстрата или растворитель, переходить от SN2-реакций и SNl-реакциям, рассмотренным в предыдущей главе.
|
|
Рис. XI-1. Зависимость скорости гидролиза алкилгалогенидов от их строения. |
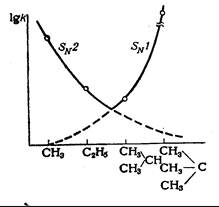
Увеличение стерического объема заместителей при реакционном центре субстрата увеличивает скорость реакции SNl и уменьшает скорость реакции SN2. Кроме того, влияние электронных эффектов заместителей в субстрате в большинстве случаев значительно сильнее проявляется в реакциях мономолекулярного замещения. Поэтому очевидно, что при переходе от первичных систем к вторичным и третичным скорость SNl-замещения должна увеличиваться, а скорость SN2 -замещения - уменьшаться, что может привести к смене механизма реакции. На рис. XI-l представлена зависимость скорости гидролиза алкилгалогенидов от строения алкильной группы. Левая часть кривой отвечает преимущественно бимолекулярному процессу, поэтому при увеличении числа алкильныx заместителей скорость процесса падает. При переходе к третичному алкилгалогениду происходит резкое увеличение скорости процесса, так как реакция начинает преимущественно идти по SN1-механизму.
Большое значение имеет природа нуклеофила и растворителя. При использовании высоко реакционноспособных нуклеофилов и апротонных биполярных растворителей (что должно способствовать реакции бимолекулярного замещения) иногда удается даже для третичных соединений осуществить SN2-механизм.
Наиболее сложно решить, по какому механизму протекает реакция в промежуточной области, где скорости процессов SN1 и SN2 соизмеримы. Можно высказать два предположения:
1) реакция одновременно идет по двум путям, и оба механизма конкурируют друг с другом;
2) реакция осуществляется по некоторому промежуточному (borderline) механизму.
В литературе имеются примеры того, что возможно одновременное течение SN1- и SN2-процессов. Например, было показано, что следующая реакция
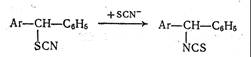 |
описывается кинетическим уравнением: υнабл = k1 [RSCN] + k2 [RSCN][SCN-] где k1 не зависит от концентрации нуклеофила.
Такая картина согласуется с одновременным образованием конечного продукта за счет как изомеризации в ионной паре или свободных ионах, что характеризуется скоростью υ(SN1) = k1[R-SCN] так и бимолекулярной реакции замещения со скоростью υ(SN2) = k2 [R-SСN][NаSСN]
Изучение замещенных соединений показало, что для мономолекулярного вклада ρ = -4,5 (корреляция с σ+), а зависимость бимолекулярной реакции от заместителей в ароматическом ядре очень слаба. Это также согласуется с одновременным течением реакций SN1 и SN2.