Вода
Харрис и Олдер (1953) в свеем расчете диэлектрической постоянной воды и других полярных жидкостей воспользовались методом Кирквуда. Однако они исключили из рассмотрения искажения поляризуемости молекул в процессе взаимодействия и предположили, что действие внешнего поля на сферический образец диэлектрика в вакууме не сопровождается изменением поляризуемости, как это получилось у Кирквуда, а состоит лишь в изменении ориентации постоянных диполей. При этом если положить постоянный дипольный момент молекулы диэлектрика равным нулю, то поляризация должна удовлетворять уравнению Клаузиуса-Моссоти. Их соотношение для ![]() имеет вид
имеет вид
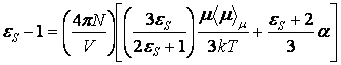 .
.
Если ![]() = 0, то получаем
= 0, то получаем
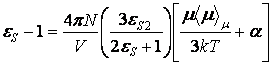 ,
,
или
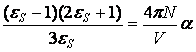 .
.
|
Таблица 4 | ||||||
| Данные для | ||||||
| Н2О | ||||||
| Т, ºС | g |
1939) |
1960) |
Олдер,1953) |
Миллер,1961) | |
| 0,0 | 2,60 | 71,9 | 73,0 | 90,7 | 88,2 | |
| 25,0 | 2,55 | 63,8 | 64,0 | 79.9 | 78,5 | |
| 62,0 | 2,49 | 53,0 | 53,0 | 65,7 | 66,4 | |
| 83,0 | 2,46 | 47,8 | 48,0 | 59,6 | 60,4 | |
| D2О | ||||||
|
Т, ºС |
|
| ||||
| 5,0 | 87,9 | 85,9 | ||||
| 25,0 | 79,5 | 78,25 | ||||
| 62,0 | 65,5 | 66,1 | ||||
| 83,0 | 59,4 | 60,1 | ||||
Оценку ![]() Харрис и Олдер заимствовали у Попла и получили для воды замечательное (в пределах 3%) согласие абсолютных значений с эксериментом и правильную температурную зависимость (табл.4). Они произвели расчет
Харрис и Олдер заимствовали у Попла и получили для воды замечательное (в пределах 3%) согласие абсолютных значений с эксериментом и правильную температурную зависимость (табл.4). Они произвели расчет ![]() и для тяжелой воды D2O c той же константой изгибания водородныйх связей
и для тяжелой воды D2O c той же константой изгибания водородныйх связей ![]() , с тем же числом соседей и тем же молярным объемом, как в обычной воде. Они учли только различную поляризуемость свободных молекул Н2О и D2O. Согласие при этих предположениях эксперимента и расчета оказалось весьма хорошим.
, с тем же числом соседей и тем же молярным объемом, как в обычной воде. Они учли только различную поляризуемость свободных молекул Н2О и D2O. Согласие при этих предположениях эксперимента и расчета оказалось весьма хорошим.
Таким образом, результаты анализа данных относительно величины и температурной зависимости ![]() в жидкой воде показывают, что основную роль в
в жидкой воде показывают, что основную роль в 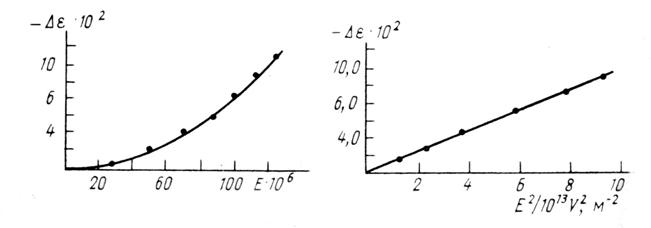 межмолекулярном взаимодействии в воде играет деформация водородных связей в жидкой воде по сравнению со льдом I.
межмолекулярном взаимодействии в воде играет деформация водородных связей в жидкой воде по сравнению со льдом I.
 Значение g для воды больше 1 при всех температурах и давлениях. Это означает, что локальное взаимодействие посредством водородных связей пяти молекул воды обусловливает суммацию дипольных моментов (g бывает меньше единицы, если молекулы какой-либо жидкости ассоциированы в пары или кольца).
Значение g для воды больше 1 при всех температурах и давлениях. Это означает, что локальное взаимодействие посредством водородных связей пяти молекул воды обусловливает суммацию дипольных моментов (g бывает меньше единицы, если молекулы какой-либо жидкости ассоциированы в пары или кольца).
Очевидно, что с увеличением напряженности внешнего электрического поля внешнее поле будет конкурировать с внутренним полем, что должно проявиться в изменении g.
![]()
Это изменение можно представить чисто формальным разложением g по степеням Е. Нечетные степени не включаются в разложение, так как g (-E) должно быть равно g (Е). Зависимость ![]() от напряженности поля Е представлена на рис.14 (Джонс, 1974).
от напряженности поля Е представлена на рис.14 (Джонс, 1974).
Начиная с напряженностей Е•105 см-1, ![]() зависит от поля нелинейно. Это означает, что внешнее поле Е•106В∙см-1 уже конкурирует с водородными связями и уменьшает g.
зависит от поля нелинейно. Это означает, что внешнее поле Е•106В∙см-1 уже конкурирует с водородными связями и уменьшает g.
Так как большинство изменений выполняется в полях меньших напряженностей, то эффект изменения g от Е можно не учитывать.